达玛烷型三萜皂苷类化合物及其制备方法和在制备抗炎药物中的应用与流程
本发明涉及化学合成,具体为从青钱柳叶中提取分离达玛烷型三萜皂苷类化合物及其制备方法和用途。
背景技术:
青钱柳为胡桃科青钱柳[cyclocaryapaliurus(batal)iljinsk(juglandaceae)]属植物,性味辛、微苦、平,具有祛风止痒的功效,用于治疗皮肤癣,产于江西、广西、贵州、湖南、湖北、四川、福建、江西、浙江、安徽等地,用药历史悠久,为我国常用中药材。
青钱柳的主要化学成分为三萜类化合物,主要包括达玛烷型三萜皂苷、黄酮类、酚酸类化合物等。现代药理学研究表明,青钱柳粗提物具有降血糖、降血压、降血脂、抗炎、抗肿瘤、抗氧化、抑菌等作用。
巨噬细胞是机体内重要的一种免疫细胞,是体内启动炎症介质产生的主要细胞,并且具有抗感染、抗肿瘤和免疫调节的作用。巨噬细胞能够被多种炎症性因子所激活,如细胞因子、细菌脂多糖lps、细胞外基质蛋白以及其他化学介质等。lps是十分重要的致炎因子,它能够刺激体内的巨噬细胞合成和释放多种内源性活性因子,进而诱发炎症。利用lps处理巨噬细胞是常用的体外炎症模型造模手段。
目前临床上常用的抗炎药主要有两类:非甾体抗炎药和甾体类抗炎药。虽然这两类抗炎药都有很好的临床抗炎效果,但长期大量使用会产生一系列不良反应和耐受性,如胃粘膜损伤、肝脏损伤、肾脏损害等。为解决药物的耐受性及不良反应,寻找新的抗炎药物以及相关新颖骨架类型的药物成为抗炎药物研究领域的热点。
天然产物是药物先导化合物的重要来源,代谢产物丰富多样,一直是药物筛选的重要源头。植物的次级代谢产物具有广泛的生理活性,如抗菌、抗肿瘤、免疫调节、抗炎、酶抑制等多种活性。一直以来,从药用植物中寻找新的药源分子都是国内外研究的热点。
技术实现要素:
本发明的目的是针对现有技术的不足,而提供一种达玛烷型三萜皂苷类化合物及其制备方法和在制备抗炎药物中的应用。用这种方法制备的该类新化合物具有抑制lps诱导的raw264.7细胞中no、tnf-α、pge2和il-6的释放,能降低inos、cox-2和nf-κb/p65蛋白的表达,显示良好的抗炎活性,可用于开发抗炎的相关药物。
实现本发明目的的技术方案是:
一种达玛烷型三萜皂苷类化合物,与现有技术不同处在于,所述达玛烷型三萜皂苷类化合物的结构如式(i)所示:
上述达玛烷型三萜皂苷类化合物的制备方法,包括如下步骤:
s1.将青钱柳叶用醇溶剂提取,其中,所述青钱柳叶与醇溶剂的质量比为1:(10-15),得到浸膏;
s2.将步骤s1制备的浸膏依次采用硅胶柱色谱、mci凝胶柱色谱、反相柱色谱、凝胶柱色谱和制备型高效液相色谱进行分离,得到式(i)所示的化合物。
优选的,所述硅胶柱色谱所用的洗脱液为石油醚、乙酸乙酯、二氯甲烷、三氯甲烷、丙酮、乙醇或甲醇中的一种溶剂或不同种溶剂的混合夜。
优选的,所述mci凝胶柱色谱所用的洗脱液为醇溶剂。
优选的,所述反相柱色谱采用的仪器为低压制备色谱、中压制备色谱、高效液相色谱或动态轴向压缩色谱,其中,反相色谱所用的流动相为有机溶剂的水溶液,有机溶剂为甲醇或乙腈。
优选的,所述反相柱色谱的洗脱为梯度洗脱,以体积分数计,所述梯度洗脱程度为:0-20min,10%-30%有机溶剂的水溶液;20-60min,30%-60%有机溶剂的水溶液;60-80min,60%-90%有机溶剂的水溶液;80-100min,100%有机溶剂的水溶液。
优选的,所述凝胶柱色谱中的凝胶为sephadexlh-20、sephadexg15或sephadexg50。
优选的,制备型高效液相色谱的流动相为甲醇-水溶液或乙腈-水溶液。
上述方法所制备的达玛烷型三萜皂苷类化合物的一种或几种在制备抗炎类药物中的应用。
上述技术方案提供了一种药物制剂,包括上述的达玛烷型三萜皂苷类化合物的一种或几种与药学上可接受的载体。
用这种方法制备的该类新化合物具有抑制lps诱导的raw264.7细胞中no、tnf-α、pge2和il-6的释放,能降低inos、cox-2和nf-κb/p65蛋白的表达,显示良好的抗炎活性,可用于开发抗炎的相关药物。
附图说明
图1为实施例中化合物it-1、2、3、4、5和6对lps刺激的raw264.7细胞中tnf-α产生的影响示意图;
图2为实施例中化合物it-1、2、3、4、5和6对lps刺激的raw264.7细胞中pge2产生的影响示意图;
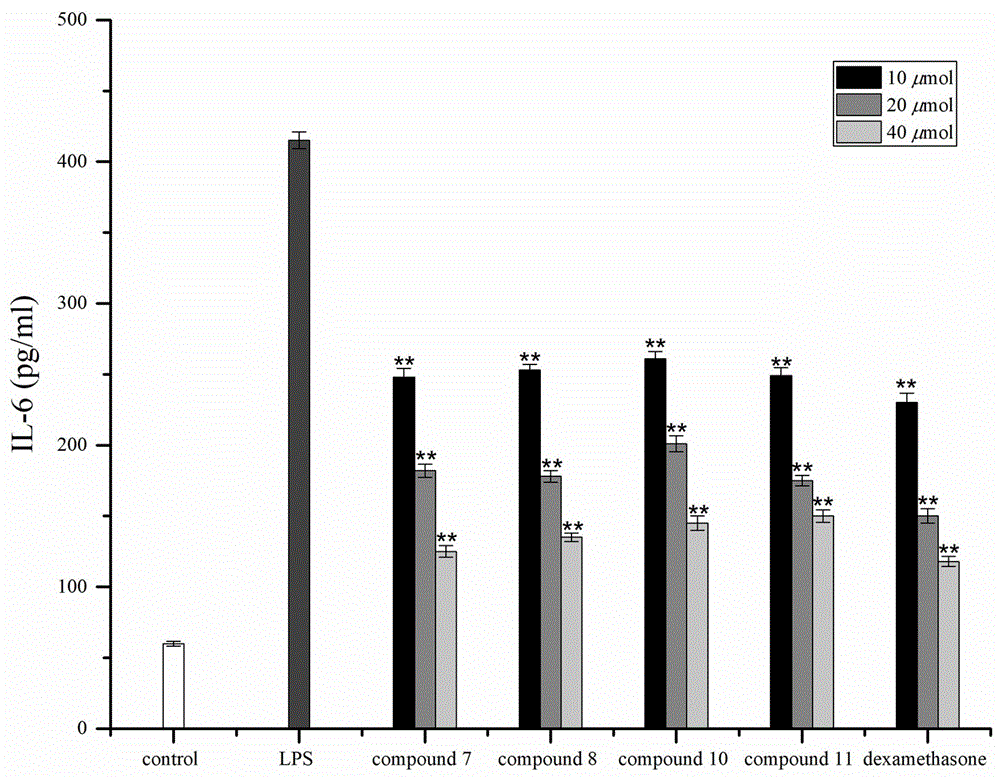
图3为实施例中化合物7、8、10和11对lps刺激的raw264.7细胞中il-6产生的影响示意图;
图4为实施例中化合物7在5、10、20μm浓度下对脂多糖诱导的raw264.7细胞中inos、cox-2和nf-κb/p65表达的影响示意图。
具体实施方式
下面结合实施例对本发明的内容作进一步的阐述,但不是对本发明的限定。
实施例:
一种达玛烷型三萜皂苷类化合物,所述达玛烷型三萜皂苷类化合物的结构如式(i)所示:
上述达玛烷型三萜皂苷类化合物的制备方法,包括如下步骤:
s1.将青钱柳叶用醇溶剂提取,其中,所述青钱柳叶与醇溶剂的质量比为1:(10-15),得到浸膏,本例中青钱柳叶与醇溶剂的质量比为1:(12-14),醇溶剂为体积分数为50%-95%的乙醇;
s2.将步骤s1制备的浸膏依次采用硅胶柱色谱、mci凝胶柱色谱、反相柱色谱、凝胶柱色谱和制备型高效液相色谱进行分离,得到式(i)所示的化合物。
优选的,所述硅胶柱色谱所用的洗脱液为石油醚、乙酸乙酯、二氯甲烷、三氯甲烷、丙酮、乙醇或甲醇中的一种溶剂或不同种溶剂的混合夜;
优选的,所述mci凝胶柱色谱所用的洗脱液为醇溶剂。
优选的,所述反相柱色谱采用的仪器为低压制备色谱、中压制备色谱、高效液相色谱或动态轴向压缩色谱,其中,反相色谱所用的流动相为有机溶剂的水溶液,有机溶剂为甲醇或乙腈。
优选的,所述反相柱色谱的洗脱为梯度洗脱,以体积分数计,所述梯度洗脱程度为:0-20min,10%-30%有机溶剂的水溶液;20-60min,30%-60%有机溶剂的水溶液;60-80min,60%-90%有机溶剂的水溶液;80-100min,100%有机溶剂的水溶液。
优选的,所述凝胶柱色谱中的凝胶为sephadexlh-20、sephadexg15或sephadexg50。
优选的,制备型高效液相色谱的流动相为甲醇-水溶液或乙腈-水溶液。
本例中,上述方法所制备的式(i)所示的达玛烷型三萜皂苷类化合物为白色粉末,化合物的紫外(uv)、核磁氢谱(1hnmr)、碳谱(13cnmr)、质谱hresims数据如表1-表3,其中表1为化合物1-6的氢谱(1hnmr)数据(500mhz,pyridine-d6),表2为化合物7-11的氢谱(1hnmr)数据(500mhz,pyridine-d6),表3为化合物1-11的碳谱(13cnmr)数据(125mhz,pyridine-d6):
化合物1:白色无定形粉末,
化合物2:白色无定形粉末,
化合物3:白色无定形粉末,
化合物4:白色无定形粉末,
化合物5:白色无定形粉末,
化合物6:白色无定形粉末,
化合物7.白色无定形粉末,
化合物8.白色无定形粉末,
化合物9:白色无定形粉末,
化合物10:白色无定形粉末,
化合物11:白色无定形粉末,
对本实施例分离得到的化合物进行结构鉴定,最终确定结构如式(i)所示,为达玛烷型三萜皂苷类化合物,所有的氢信号归属见表1和表2,碳信号归属见表3。
上述方法所制备的达玛烷型三萜皂苷类化合物的一种或几种在制备抗炎类药物中的应用。
上述技术方案提供了一种药物制剂,包括上述的达玛烷型三萜皂苷类化合物的一种或几种与药学上可接受的载体。
所述药学上可接受的载体可根据药剂学领域的常用辅料,根据剂型和实际情况进行恰当选择,例如常用的辅料有淀粉、低取代羟丙基纤维素、微粉硅胶、硬脂酸镁、淀粉浆、蔗糖、糊精、羧甲基淀粉钠、滑石粉、聚山梨酯、聚乙二醇、注射用大豆磷脂和注射用甘油等;采用本实施例得到的式(i)所示的达玛烷型三萜皂苷类化合物的一种或几种制备所需药物的各种剂型时,可以按照药剂学领域的常规生产方法制备,如将该提取物与一种或多种载体混合,然后制成相应的剂型,中药制剂的剂型包括注射剂、片剂、栓剂、软膏剂、凝胶剂、丸剂、片剂、颗粒剂、胶囊剂和合剂。
本实施例通过对青钱柳叶进行提取,得到浸膏;然后将得到的浸膏进行分离,选择特定出峰时间的化合物,即得到式(i)所示的达玛烷型三萜皂苷类化合物的一种或几种,且通过细胞实验发现,该类达玛烷型三萜皂苷类化合物能够极显著的抑制lps诱导的no、tnf-α、pge2和il-6的释放,降低inos、cox-2和nf-κb/p65的表达,显示极为显著的抗炎活性。
具体地:
实施例1:
制备如式(i)所示的结构(1-11)的达玛烷型三萜皂苷类化合物的方法,包括如下步骤:
1-1)将青钱柳叶用12倍量的50%乙醇在加热提取3次,每次2h,过滤,将滤液浓缩、回收乙醇得到粗浸膏;
1-2)取步骤1-1)中得到的粗浸膏与80-100目硅胶以质量比1:1.4的比例进行拌样,经硅胶柱色谱分离,采用二氯甲烷-甲醇混合溶液进行梯度洗脱,所述二氯甲烷-甲醇体积比为(100:0-0:100),洗脱出的流份用薄层色谱分析合并,收集二氯甲烷-甲醇体积比为(10-5):1洗脱部位,得硅胶柱色谱分离后的粗品;
1-3)将步骤1-2)中得到的硅胶柱色谱分离后的粗品,经反相c18动态轴向压缩柱色谱进行分离,洗脱程序为0-20min,10%-30%有机溶剂的水溶液;20-60min,30%-60%有机溶剂的水溶液;60-80min,60%-90%有机溶剂的水溶液;80-100min,100%乙腈-水溶液,收集35~60min洗脱部位,得反相柱色谱分离后的粗品;
1-4)将步骤1-3)中用c18反相柱色谱分离后的粗品,经sephadexlh-20凝胶柱色谱分离,以二氯甲烷:甲醇(1:1)进行纯化,得到纯化后的样品;
1-5)将步骤1-4)中得到的纯化后的样品,通过制备型高效液相色谱进行分离,进行c18反相柱色谱(21.2×250mm,5μm);以甲醇和水以及乙腈和水的混合溶液为流动相进行洗脱,流速8ml/min;检测波长为205nm,收集洗脱液减压干燥,得到式(i)所示的达玛烷型三萜皂苷类1-11。
用紫外(uv)、一维核磁共振谱(1hnmr和13cnmr)、二维核磁共振谱(hsqc,hmbc,noesy和tocsy)、质谱(ms)等手段,并结合参考文献,对分离得到的单体化合物进行结构鉴定,得到如式(i)所示的结构(1-11),这些化合物是新的达玛烷型三萜皂苷类,所有化合物的氢信号归属见表1和表2,碳信号归属见表3。
实施例2:
制备如式(i)所示的结构(1-11)的达玛烷型三萜皂苷类化合物的方法,包括如下步骤:
2-1)将青钱柳叶用12倍量的50%乙醇在加热提取3次,每次2h,过滤,将滤液浓缩、回收乙醇得到浸膏,将粗浸膏悬浮于水中,先用石油醚萃取,在用乙酸乙酯萃取,回收溶剂,得到萃取物;
2-2)取步骤2-1)中得到的粗浸膏与80-100目硅胶以质量比1:1.3的比例进行拌样,经硅胶柱色谱分离,采用二氯甲烷-甲醇混合溶液进行梯度洗脱;所述二氯甲烷-甲醇体积比为(100:0-0:100),洗脱出的流份用薄层色谱分析合并,收集二氯甲烷-甲醇体积比为(10-5):1洗脱部位,得硅胶柱色谱分离后的粗品;
2-3)将步骤2.2中得到的硅胶柱色谱分离后的粗品,经反相c18动态轴向压缩柱色谱进行分离,洗脱程序为0-20min,10%-30%有机溶剂的水溶液;20-60min,30%-60%有机溶剂的水溶液;60-80min,60%-90%有机溶剂的水溶液;80-100min,100%乙腈-水溶液,收集35-60min洗脱部位,得反相柱色谱分离后的粗品;
2-4)将步骤2-3)中用c18反相柱色谱分离后的粗品,经sephadexlh-20凝胶柱色谱分离,以二氯甲烷:甲醇(1:1)进行纯化,得到纯化后的样品;
2-5)将步骤2-4)中得到的纯化后的样品,通过制备型高效液相色谱进行分离,进行c18反相柱色谱(21.2×250mm,5μm);以甲醇和水以及乙腈和水的混合溶液为流动相进行洗脱,流速8ml/min;检测波长为205nm,收集洗脱液减压干燥,得到式(i)所示的达玛烷型三萜皂苷类化合物1-11。
实施例3:
制备如式(i)所示的结构(1-11)的达玛烷型三萜皂苷类化合物的方法,包括如下步骤:
3-1)将青钱柳叶用13倍量的75%乙醇在加热提取3次,每次3h,过滤,将滤液浓缩、回收乙醇得到浸膏;
3-2)取步骤3-1)中得到的粗浸膏与80-100目硅胶以质量比1:1.3的比例进行拌样,经硅胶柱色谱分离,采用二氯甲烷-甲醇混合溶液进行梯度洗脱;所述二氯甲烷-甲醇体积比为(100:0-0:100),洗脱出的流份用薄层色谱分析合并,收集二氯甲烷-甲醇体积比为(10-5):1洗脱部位,得硅胶柱色谱分离后的粗品;
3-3)将步骤3-2)中得到的硅胶柱色谱分离后的粗品,经反相c18动态轴向压缩柱色谱进行分离,洗脱程序为0-20min,10%-30%有机溶剂的水溶液;20-60min,30%-60%有机溶剂的水溶液;60-80min,60%-90%有机溶剂的水溶液;80-100min,100%乙腈-水溶液,收集35-60min洗脱部位,得反相柱色谱分离后的粗品;
3-4)将步骤3-3)中用c18反相柱色谱分离后的粗品,经sephadexlh-20凝胶柱色谱分离,以二氯甲烷:甲醇(1:1)进行纯化,得到纯化后的样品;
3-5)将步骤3-4)中得到的纯化后的样品,通过制备型高效液相色谱进行分离,进行c18反相柱色谱(21.2×250mm,5μm);以甲醇和水以及乙腈和水的混合溶液为流动相进行洗脱,流速8ml/min;检测波长为205nm,收集洗脱液减压干燥,得到式(i)所示的达玛烷型三萜皂苷类化合物1-11。
实施例4:
4-1)将青钱柳叶用14倍量的95%乙醇在加热提取3次,每次1.5h,过滤,将滤液浓缩、回收乙醇得到浸膏;
4-2)取步骤4-1)中得到的粗浸膏与80-100目硅胶以质量比1:1.3的比例进行拌样,经硅胶柱色谱分离,采用二氯甲烷-甲醇混合溶液进行梯度洗脱;所述二氯甲烷-甲醇体积比为(100:0-0:100),洗脱出的流份用薄层色谱分析合并,收集二氯甲烷-甲醇体积比为(10-5):1洗脱部位,得硅胶柱色谱分离后的粗品;
4-3)将步骤4-2)中得到的硅胶柱色谱分离后的粗品,经反相c18动态轴向压缩柱色谱进行分离,洗脱程序为0-20min,10%-30%有机溶剂的水溶液;20-60min,30%-60%有机溶剂的水溶液;60-80min,60%-90%有机溶剂的水溶液;80-100min,100%乙腈-水溶液,收集35-60min洗脱部位,得反相柱色谱分离后的粗品;
4-4)将步骤4-1)中用c18反相柱色谱分离后的粗品,经sephadexlh-20凝胶柱色谱分离,以二氯甲烷:甲醇(1:1)进行纯化,得到纯化后的样品;
4-5)将步骤4-4)中得到的纯化后的样品,通过制备型高效液相色谱进行分离,进行c18反相柱色谱(21.2×250mm,5μm);以甲醇和水以及乙腈和水的混合溶液为流动相进行洗脱,流速8ml/min;检测波长为205nm,收集洗脱液减压干燥,得到式(i)所示的达玛烷型三萜皂苷类化合物1-11。
实施例5:
对一种上述式(i)所示的达玛烷型三萜皂苷类化合物1-11的抗炎活性研究验证:
5-1)抗炎活性实验,包括:
5-1-1)实验试剂及仪器:
s1)主要试剂:巨噬细胞raw264.7(购于中国科学院生化细胞所),dmem高糖培养基(hyclone公司,美国),新生牛血清(hyclone公司,美国),lps、mtt(sigma公司,美国),notestkit(碧云天生物技术研究所),rabbitanti-inos、rabbitanti-cox-2、rabbitanti-nf-κb(abcam公司,英国),elisatestkit(武汉伊莱瑞特);
s2)主要仪器:infinitem1000酶标仪(瑞士tecan),co2培养箱(美国thermo),96孔细胞培养板(美国corning,0-10μl,0-200μl,1ml移液器(德国eppendorf),台式微量离心机(美国thermo),倒置显微镜(日本olympus)。
5-2)实验方法与步骤,包括:
5-2-1)细胞毒性实验:
5-2-1-1)细胞抗炎实验分组:
将实验分为:①空白组:raw264.7细胞不加入任何药物;②lps组:细胞+5ng/ml的lps溶液;③加药组:细胞+5ng/ml的lps溶液+不同浓度阳性对照或化合物;
5-2-1-2)mtt实验方法:①细胞培养:将raw264.7细胞进行消化,进行细胞计数,将细胞浓度调整至1×105个/ml;②种板:按照180μl/孔的细胞悬液接种于96孔板中,在37℃孵育24小时;③加lps:换新鲜培养基180μl,在每孔加入10μl的浓度为5ng/ml的lps溶液,在37℃孵育2个小时;④加药:把不同浓度的阳性对照和不同浓度的化合物,各10μl加入孔板,每个样设定三个复孔,于37℃培养24个小时;⑤加mtt:每孔加mtt溶液,37℃培养4个小时;⑥加dmso:弃掉上清液,每孔加入100μldmso,震荡15min,完全溶解甲臜;⑦测od值:将96孔板置于酶标仪中,选择波长为490nm,测定od;⑧计算:计算细胞存活率;
5-2-2)griess法测lps诱导raw264.7巨噬细胞no的含量,elisa法测tnf-α、pge2和il-6的含量:①raw264.7细胞的培养,与步骤5-2-1-2相同,96孔板换成24孔板,细胞加入体积和加药量体积比原来扩大4倍即可;②加药:对加药组按照要求进行加药,一一标识;③取样:收集上清液于离心管中,离心、进行下一步试剂盒操作;④no的测试步骤按试剂盒说明书进行,最后根据抑制率和浓度的关系计算ic50值:
no抑制率(%)=[(alps组-a药物组)/(alps组-a空白组)]×100%;
化合物抑制raw264.7细胞no释放的结果见表4。从表4看到,化合物7、8、10和11抑制raw264.7细胞no的释放,化合物7抑制no释放的活性与阳性对照地塞米松(dexamethasone)一样,表明青钱柳中含有抗炎活性良好的活性达玛烷型三萜皂苷类化合物。
表4.no释放抑制活性
⑤elisa试剂盒tnf-α、pge2和il-6操作说明来一一进行操作,测定这些因子的含量,所有数值是三次测定的平均值±se。**p<0.01与lps处理的细胞相比;
5-2-3)western-blot法测定细胞中inos、cox-2和nf-κb/p65蛋白的表达:
s1)蛋白样品制备和蛋白定量:①细胞培养与步骤5-2-1-2相同;②提取细胞蛋白收集细胞,加细胞裂解液,离心,收集细胞蛋白;③bca(喹啉酸)法蛋白进行定量,上样蛋白量的确定;
s2)蛋白质变性:加上样缓冲液,煮沸,置到冰上冷却;备用(3)sds-page电泳:①制分离胶:根据所有蛋白的分子量来确定相应分离胶的配比,制备分离胶;②加浓缩胶:分离胶凝固后,灌5%浓缩胶至分离胶顶端;然插入梳子;③取胶至电泳槽:浓缩胶凝固,取出梳子,将胶板从固定钳中取下,冲水,置于电泳槽,玻璃板的高端向外;④上样:按从左到右的顺序加marker和样品蛋白;⑤电泳:初设电压80v,带样品跑至分离胶部分,电压调为100v,至各组蛋白置合适位置,结束电泳;
5-2-4)转膜:在电转移仪夹子二面依次铺好海绵垫和三层滤纸,将胶转移到滤纸上,将合适大小的硝酸纤维素膜盖于胶上,盖3张滤纸,除气泡,合起夹子,装好电转移仪(注意电极对应),根据不同蛋白需要选定所需的电流和时间;
5-2-5)免疫反应:①封闭:取出pvdf膜,加入在装有10ml封闭液的玻璃皿中,在摇床上缓慢摇动2h或者4℃过夜;②孵育一抗:用tbst洗过的洗膜,膜蛋白面朝下,放于抗体液面上;室温下孵育1-2h后,用tbst在室温下摇床上洗三次,每次10min;③孵育二抗:二抗稀释液并与膜接触,孵育1-2h后,tbst与摇床洗三次,每次10min,准备进行下一步;
5-2-6)化学发光显影:凝胶图象分析:加入显影液用,将pvdf进行拍照处理;5-2-7)数据处理:用凝胶图象处理系统分析目标蛋白的净光密度值,所有数值是三次测定的平均值±se。**p<0.01与lps处理的细胞相比。
如图1所示,化合物it-1、2、3、4、5和6对lps刺激的raw264.7细胞中tnf-α产生的影响,化合物用lps(100ng/ml)2h刺激细胞,然后用化合物(10、20、40μm)处理24h,用酶联免疫吸附测定上清液中tnf-α的浓度,以地塞米松为阳性对照品,与化合物浓度相同。
如图2所示,化合物it-1、2、3、4、5和6对lps刺激的raw264.7细胞中pge2产生的影响,化合物用lps(100ng/ml)2h刺激细胞,然后用化合物(12.5,25,50μm)处理24h,用酶联免疫吸附测定上清液中tnf-α的浓度,以地塞米松为阳性对照品,与化合物浓度相同。
如图3所示,化合物7、8、10和11对lps刺激的raw264.7细胞中il-6产生的影响,化合物用lps(100ng/ml)2h刺激细胞,然后用化合物(10、20、40μm)处理24h,用酶联免疫吸附测定上清液中il-6的浓度,以地塞米松为阳性对照品,与化合物浓度相同。
如图4所示,化合物7在5、10、20μm浓度下对脂多糖诱导的raw264.7细胞中inos、cox-2和nf-κb/p65表达的影响,β-actin做内参。
本实施例制备的化合物具有抑制lps诱导的raw264.7细胞中no、tnf-α、pge2和il-6的释放,显著降低inos、cox-2和nf-κb/p65蛋白的表达,显示良好的抗炎活性。
实施例6:
上述式(i)所示的达玛烷型三萜皂苷类化合物的一种或几种制备片剂药物:
将4g式(i)所示结构的化合物一种或几种和100g淀粉、15g羧甲基淀粉钠、1.6g滑石粉、100g糊精、1.6g硬脂酸镁及适量10%淀粉浆适混合,按照常规方法制成式i所示结构的化合物片剂2000片,服用:每日3次,每次1片。
实施例7:
上述式(i)所示的达玛烷型三萜皂苷类化合物的一种或几种制备丸剂药物:
将4g式i所示结构的化合物和24g聚乙二醇-6000、160g聚山梨酯-80、适量液状石蜡混合,按照常规方法制成式i所示结构的化合物丸剂2000粒,服用:每日3次,每次1粒。
实施例8:
上述式(i)所示的达玛烷型三萜皂苷类化合物的一种或几种制备注射剂药物:
将3g式i所示结构的化合物和30g注射用大豆磷脂、50g注射用甘油,注射用水定容至2000ml,按照常规方法制成式i所示结构的化合物注射剂2000支,服用:每日1次,每次1支,至少采用250ml5%葡萄糖注射液稀释后静脉滴注。
- 还没有人留言评论。精彩留言会获得点赞!