一种二丙酸倍他米松的制备方法与流程
本发明属于化学合成
技术领域:
,具体涉及一种二丙酸倍他米松的制备方法。
背景技术:
:二丙酸倍他米松(betamethasonedipropionate)又名倍他米松双丙酸酯,化学名为16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯,是糖皮质激素类药物倍他米松的前药。临床上主要用于治疗皮肤炎症和瘙痒症。二丙酸倍他米松的结构式如式ⅰ所示:二丙酸倍他米松传统的合成方法,用倍他米松与原丙酸三乙酯环合反应得到倍他米松-17,21-原丙酸环酯,然后21位选择性水解得到倍他米松17-丙酸酯,最后21位羟基进行丙酰化得到目标物。传统的合成方法中,因倍他米松17-丙酸酯在大多数溶剂中难溶的特性,第二步常采用四氢呋喃、2-甲基四氢呋喃、吡啶或三氯甲烷等溶剂,用大量的吡啶或三乙胺做催化剂反应,所用溶剂或试剂毒性较大。(张宇松;金炜华;汪卫军;二丙酸倍他米松的合成研究[j];中国现代应用药学;2011年07期)采用2-甲基四氢呋喃做溶剂,用大量的三乙胺做催化剂。反应后产生大量的废液,对环境污染严重,无法适应目前的环保要求,且2-甲基四氢呋喃价格昂贵。用乙酸乙酯/石油醚精制除杂效果较差,收率低,不适合商业化生产。因此,本发明提出了一种简化的合成方法,并克服了环境污染问题和成本问题。技术实现要素:有鉴于此,本发明的目的在于提供一种二丙酸倍他米松(16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯)的制备方法,本发明方法采用了成本交底且环保的溶剂和催化剂,且收率高,纯度高,适合商业化生产。为实现上述目的,本发明采用以下方案:所述方法为以式ⅱ所示的化合物(倍他米松-17-丙酸酯)为原料和丙酸酐为原料,在4-二甲氨基吡啶做催化剂(dmap)的条件下,反应生成式ⅰ所示的化合物,并采用无水乙醇、二氯甲烷和正己烷精制得到二丙酸倍他米松,反应式如下:进一步,所述方法具体为如下步骤:1)反应:氮气保护下,将二氯甲烷加入反应釜中搅拌,加入式ⅱ所示的化合物、催化剂4-二甲氨基吡啶,降温,加入丙酸酐与二氯甲烷的溶液反应;2)干燥浓缩:反应后加水淬灭,静置分层,水层弃去,有机层用水洗涤,加无水硫酸钠干燥,过滤,收集滤液,减压浓缩,浓缩后加入正己烷,继续浓缩至断流,再加入正己烷,浓缩至断流;3)精制:加入二氯甲烷、无水乙醇后,加热溶解,滴加正己烷,降温析晶,离心,用二氯甲烷与正己烷混合液洗涤,滤饼真空干燥得所述二丙酸倍他米松。进一步,步骤1)反应的温度为-10~25℃,优选为0~10℃。更进一步,反应时间为3~5小时。进一步,步骤2)浓缩的温度为40℃~50℃,减压的真空度-0.05mpa~-0.10mpa。进一步,步骤3)无水乙醇与步骤2)得到的粗品式ⅰ所示的化合物的质量比为0.1%~20%,优选为0.1%~5%,更优选为5%。具体的,粗品式ⅰ的量按投式ⅱ化合物反应后生成式ⅰ化合物的理论量计。进一步,步骤3)二氯甲烷与步骤2)得到的粗品式ⅰ所示的化合物的体积比为1~5,优选为1~3,更优选为1~2。进一步,步骤3)正己烷(精制时采用的量)与步骤2)得到的粗品式ⅰ所示的化合物的体积比为1~20,优选为1~5,更优选为1~4。进一步,步骤3)降温析晶的温度为0~5℃,滤饼真空干燥的温度为45℃~55℃,真空度-0.08mpa~-0.10mpa。进一步,对得到的二丙酸倍他米松进行hplc测定纯度,柱温20℃,流速0.2ml/min,以乙腈水溶液作为流动相,采用等度洗脱。本发明的有益效果在于:1)本发明的制备工艺简单,反应温度适中,无超低温或高温反应,便于工业化生产;2)制备中所用溶剂或试剂便宜易得,加料方便,运输储存简单;3)反应催化剂用量少,收率高,产生的废液少,对环境友好;4)精制溶剂体系除杂能力强,收率高,适合商业化生产。附图说明图1为实施例1二丙酸倍他米松的hplc图。图2为实施例2二丙酸倍他米松的hplc图。图3为实施例3二丙酸倍他米松的hplc图。图4为实施例4二丙酸倍他米松的hplc图。图5为实施例5二丙酸倍他米松的hplc图。图6为实施例6二丙酸倍他米松的hplc图。具体实施方式所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述
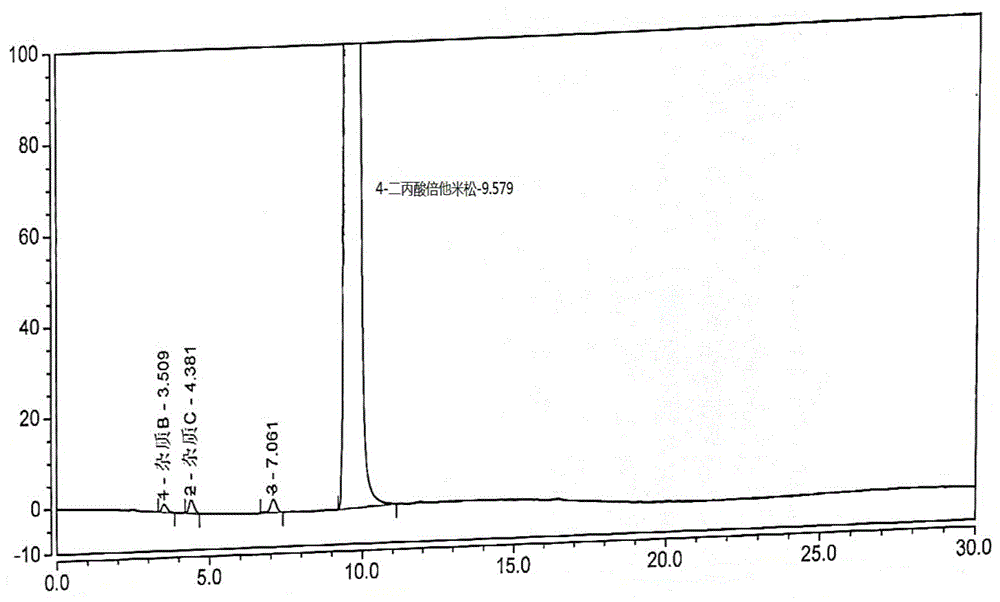
发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。高效液相色谱法(hplc)测定条件色谱柱:十八烷基硅烷键合硅胶为填充剂(phenomenexlunac18(2)-hst2mm×100mm,2.5μm)。洗脱方式:等度洗脱。流动相:35ml水与56ml乙腈混匀放置室温后,加水稀释至100ml。检测波长:254nm。柱温:20℃。流速:0.2ml/min。进样量:5μl。配样浓度:2.4mg/ml(乙腈作溶剂)。实施例1二丙酸倍他米松的制备氮气保护下,将二氯甲烷(14.4kg)加入反应釜中,开启搅拌,加入倍他米松-17-丙酸酯(式ⅱ化合物,0.90kg,2.0mol,1.0eq)、dmap(催化剂)(61.3g,0.5mol,0.25eq)。降温至0~10℃,加入丙酸酐(313g,2.4mol,1.2eq)与二氯甲烷(1.197kg)的溶液,反应3~5小时后,加入水(4.5kg)淬灭,静置分层,水层弃去,有机层用水洗涤3次,加无水硫酸钠(0.27kg)干燥,过滤,收集滤液,减压浓缩(温度40℃~50℃,真空度-0.05mpa~-0.10mpa),浓缩至剩余少量液体后加入正己烷(0.594kg),继续浓缩至断流;加入二氯甲烷(2.68kg),无水乙醇(50.4ml),加热溶解,滴加正己烷(2.66kg),降温至0~5℃搅拌析晶,离心,用二氯甲烷与正己烷混合液洗涤,滤饼于50℃(45℃~55℃),真空-0.09mpa(-0.08mpa~-0.10mpa)干燥得16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯(0.97kg),收率96%,hplc纯度99.80%(见附图1),积分结果如下表所示。实施例2二丙酸倍他米松的制备氮气保护下,将二氯甲烷(493ml)加入反应釜中,开启搅拌,加入倍他米松-17-丙酸酯(式ⅱ化合物,44.85g,0.1mol,1.0eq)、dmap(1.2g,0.01mol,0.1eq)。降温至15~25℃,加入丙酸酐(15.6g,0.12mol,1.2eq)与二氯甲烷(45ml)的溶液,反应3~5小时后,加入水(250ml)淬灭,静置分层,水层弃去,有机层用水洗涤3次,加无水硫酸钠(25g)干燥,过滤,收集滤液,减压浓缩(温度40℃~50℃,真空度-0.05mpa~-0.10mpa),浓缩至剩余少量液体后加入正己烷(50ml),继续浓缩至断流,再加入正己烷(50ml),浓缩至断流;加入二氯甲烷(101ml),无水乙醇(2.5ml),加热溶解,滴加正己烷(202ml),降温至0~5℃搅拌析晶,过滤,用二氯甲烷与正己烷混合液洗涤,滤饼于50℃(45℃~55℃),真空-0.09mpa(-0.08mpa~-0.10mpa)干燥得16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯(44.4g),收率88.1%,hplc纯度99.43%(见附图2),积分结果如下表所示。实施例3二丙酸倍他米松的制备氮气保护下,将二氯甲烷(493ml)加入反应釜中,开启搅拌,加入倍他米松-17-丙酸酯(式ⅱ化合物,44.85g,0.1mol,1.0eq)、dmap(6.1g,0.05mol,0.5eq)。降温至-10~0℃,加入丙酸酐(15.6g,0.12mol,1.2eq)与二氯甲烷(45ml)的溶液,反应3~5小时后,加入水(250ml)淬灭,静置分层,水层弃去,有机层用水洗涤3次,加无水硫酸钠(25g)干燥,过滤,收集滤液,减压浓缩(温度40℃~50℃,真空度-0.05mpa~-0.10mpa),浓缩至剩余少量液体后加入正己烷(50ml),继续浓缩至断流,再加入正己烷(50ml),浓缩至断流;加入二氯甲烷(101ml),无水乙醇(2.5ml),加热溶解,滴加正己烷(202ml),降温至0~5℃搅拌析晶,过滤,用二氯甲烷与正己烷混合液洗涤,滤饼于50℃(45℃~55℃),真空-0.09mpa(-0.08mpa~-0.10mpa)干燥得16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯(40.8g),收率80%,hplc纯度99.19%(见附图3),积分结果如下表所示。实施例4二丙酸倍他米松的制备氮气保护下,将二氯甲烷(986ml)加入反应釜中,开启搅拌,加入倍他米松-17-丙酸酯(式ⅱ化合物,89.7g,0.2mol,1.0eq)、dmap(6.1g,0.05mol,0.25eq)。降温至15~25℃,加入丙酸酐(31.2g,0.24mol,1.2eq)与二氯甲烷(90ml)的溶液,反应3~5小时后,加入水(500ml)淬灭,静置分层,水层弃去,有机层用水洗涤3次,加无水硫酸钠(50g)干燥,过滤,收集滤液,减压浓缩(温度40℃~50℃,真空度-0.05mpa~-0.10mpa),浓缩至剩余少量液体后加入正己烷(100ml),继续浓缩至断流,再加入正己烷(100ml),浓缩至断流;加入二氯甲烷(101ml),无水乙醇(0.1ml),加热溶解,滴加正己烷(101ml),降温至0~5℃搅拌析晶,过滤,用二氯甲烷与正己烷混合液洗涤,滤饼于50℃(45℃~55℃),真空-0.09mpa(-0.08mpa~-0.10mpa)干燥得16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯(90.7g),收率90%,hplc纯度98.98%(见附图4),积分结果如下表所示。实施例5二丙酸倍他米松的制备氮气保护下,将二氯甲烷(986ml)加入反应釜中,开启搅拌,加入倍他米松-17-丙酸酯(式ⅱ化合物,89.7g,0.2mol,1.0eq)、dmap(6.1g,0.05mol,0.25eq)。降温至15~25℃,加入丙酸酐(31.2g,0.24mol,1.2eq)与二氯甲烷(90ml)的溶液,反应3~5小时后,加入水(500ml)淬灭,静置分层,水层弃去,有机层用水洗涤3次,加无水硫酸钠(50g)干燥,过滤,收集滤液,减压浓缩(温度40℃~50℃,真空度-0.05mpa~-0.10mpa),浓缩至剩余少量液体后加入正己烷(100ml),继续浓缩至断流,再加入正己烷(100ml),浓缩至断流;加入二氯甲烷(505ml),无水乙醇(20.2ml),加热溶解,滴加正己烷(2020ml),降温至0~5℃搅拌析晶,过滤,用二氯甲烷与正己烷混合液洗涤,滤饼于50℃(45℃~55℃),真空-0.09mpa(-0.08mpa~-0.10mpa)干燥得16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯(89.7g),收率89%,hplc纯度99.80%(见附图5),积分结果如下表所示。实施例6二丙酸倍他米松的制备氮气保护下,将乙酸乙酯(31ml)加入反应釜中,开启搅拌,加入倍他米松-17-丙酸酯(式ⅱ化合物,2.5g,5.6x10-3mol,1.0eq)、吡啶(催化剂)(1.75g,00.22mol,3.95eq),加入丙酸酐(2.25g,0.017mol,3.1eq),升温至回流反应7小时后,室温搅拌过夜,减压浓缩至断流,加入乙酸乙酯2.5ml,加热至40℃搅拌溶解后,降温至室温搅拌30分钟,降温至0℃~10℃搅拌1小时,过滤,滤饼于50℃(45℃~55℃),真空-0.09mpa(-0.08mpa~-0.10mpa)干燥得16β-甲基-11β,17α,21-三羟基-9α-氟孕甾-1,4-二烯-3,20-二酮-17,21-二丙酸酯(89.7g),收率61%,hplc纯度97.29%(见附图6),积分结果如下表所示。序号保留时间峰名称峰面积mau*min峰高mau峰面积%拖尾因子分离度塔板数12.7170.1670.3660.100.8511.7317328.8000.1991.4280.120.935.282685139.9382.61718.5971.630.8610.2333519411.8350.0230.1690.01n.a.n.a.n.a.512.1420.1400.9990.090.872.4651180612.6380.0560.4850.031.057.9870416714.277156.2951077.79797.290.8615.5266626817.7220.3192.2040.20n.a.1.37100137918.0350.8305.4150.52n.a.n.a.95361总和100.00最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1