一种靛红芳烃共聚物、制备方法及应用与流程
本发明属于高分子材料及其聚合物离子交换膜领域,涉及一种靛红芳烃共聚物、制备方法及应用。
背景技术:
聚合物离子交换膜燃料电池作为清洁的氢能源技术,具有室温启动、能量效率高、零污染等优点在交通运输、航空航天、舰船以及军事科技领域具有广泛应用。目前,质子交换膜燃料电池(pemfc)运行时需要贵金属铂催化剂,使用价格昂贵的全氟聚合物离子膜如美国杜邦公司的nafion离子膜,因此pemfc成本较高,使其推广应用受到限制。碱性阴离子交换膜燃料电池(aemfc),在碱性条件下运行,可以使用非贵金属催化剂如镍、钴、银等,避免使用资源有限的贵金属铂催化剂,同时具有高效氧还原动力学,因此,aemfc技术成本较低、电池性能较高,近年来受到较大关注。
目前,在aemfc中存在的瓶颈问题之一,是碱性环境中聚合物离子膜的机械性能及化学稳定性较差,以及膜电极中电解质溶液性能不佳,导致燃料电池性能不佳,因此急需开发能满足实际应用的碱性离子膜材料及聚合物电解质溶液。olsson等合成的哌啶芳烃聚合物,其在60℃,2mnaoh溶液中可稳定15天,而氢氧根传导率在80℃时达到89ms·cm-1[olssonjsetal,advancedfunctionalmaterials,2018,28(2):1702758]。彭等合成了聚n-甲基哌啶芳烃共聚物,在氢氧燃料电池中峰值功率密度高达1.5w·cm-2,电池稳定工作达到100h[pengh,etal.journalofpowersources,2018,390:165-167]。
技术实现要素:
针对目前碱性燃料电池中聚合物离子膜存在机械性能及化学稳定性较差的关键问题,本发明提供了一种具有机械性能好、耐碱稳定性高的靛红芳烃共聚物、其电解质溶液及其离子交换膜、制备方法与应用。本发明在共聚物中引入靛红结构单元,方便聚合物进行交联及功能化,进一步提高聚合物离子膜、电解质溶液(ionomer)的机械性能和化学稳定性,在燃料电池、储能电池、电解水制氢气、膜分离及其它电化学器件等领域应用广泛。
本发明是采用以下技术方案实现的:
一种靛红芳烃共聚物,结构如通式ⅰ所示:
其中,0<n<1;
r为h或c1~c12的烷基、二溴代烷、烯烃、苄基苯乙烯、环氧基、丙烯酸酯或-(ch2)n’-g,其中g是三甲胺、n-甲基哌啶、n-甲基吡咯或n-甲基吗啉烷基化的铵离子;n’=1~10。
ar为芳烃,结构如下:
其中,r1是甲基或c1~c12的烷基。
一种靛红芳烃共聚物的制备方法,具体步骤如下:
步骤一,取代靛红的合成
将靛红溶解在溶剂a中制成1-20wt%溶液,加入过量无水k2co3搅拌溶解,再加入化合物b,控制靛红与化合物b的摩尔比为1:1~1:3,在20-60℃反应10-50h;反应结束后,将反应溶液倒入冰水中,用萃取剂萃取,旋蒸,粗产物用乙醇重结晶,得到取代靛红。
所述的溶剂a是n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、氯仿、二甲基亚砜、n-甲基吡咯烷酮中一种或两种以上混合。
所述的化合物b是溴代烷烃、溴代烯烃、乙烯基苄基氯、丙烯酸缩水甘油酯(gma)、溴代烷基铵盐br-(ch2)n’-g或1,6-二溴代烷。
所述的萃取剂为甲醇、甲苯、丙酮、石油醚、乙醚、氯仿中一种或两种以上混合。
步骤二,靛红芳烃共聚物的合成
将步骤一得到的取代靛红、n-甲基-4-哌啶酮和芳烃加入二氯甲烷中,冰浴下搅拌溶解使取代靛红的单体浓度为15-40wt%,得到混合液;向混合液中加入三氟甲磺酸和三氟乙酸作为催化剂,在0~5℃搅拌反应14~48h;反应结束,将产物倒入冰水中,过滤,产物用nahco3除去过量的酸,水洗,过滤,真空干燥,得到白色纤维状的靛红芳烃共聚物;将共聚物在溶剂a溶解,然后倒入沉淀剂中,过滤,干燥,得到精制的纤维状的靛红芳烃共聚物。
所述的取代靛红和n-甲基-4-哌啶酮的总摩尔数与芳烃摩尔数之比为(1~1.1):1;取代靛红和n-甲基-4-哌啶酮的摩尔比为1:9~4:6;所述的三氟甲磺酸与芳烃摩尔比为(6~14):1;所述的三氟甲磺酸与三氟乙酸的体积比为(8~14):1。
所述的芳烃是联苯、对三联苯、间三联苯、9,9-二烷基芴或双酚芴。
所述的沉淀剂为甲醇、乙醇、水、石油醚、乙酸乙酯、丙酮、乙醚中一种或两种以上混合。
一种靛红芳烃共聚物用于制备阴离子交换膜的方法,步骤如下:
步骤(1)、带铵阳离子的靛红芳烃共聚物的合成
将靛红芳烃共聚物在溶剂a中溶解得到5-20wt%溶液,加入碘甲烷或溴代烷r1-br,其中,碘甲烷或r1-br与靛红芳烃共聚物的摩尔比为(5~15):1,在30~100℃下反应20~100h;反应结束后,将反应液倒入沉淀剂中,过滤,水洗产物,得到带铵阳离子的靛红芳烃共聚物,其结构如通式ⅱ所示:
其中,0<n<1;x-是反离子,为br-或oh-离子;r1是甲基或c1~c12的烷基。
所述的溶剂a是n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、氯仿、二甲基亚砜、n-甲基吡咯烷酮中一种或两种以上混合。
所述的沉淀剂为甲醇、乙醇、水、石油醚、乙酸乙酯、丙酮、乙醚中一种或两种以上混合。
步骤(2)、带铵阳离子的靛红芳烃共聚物的电解质溶液(ionomer)制备
将步骤(1)制备的带铵阳离子的靛红芳烃共聚物加入溶剂a中,搅拌溶解,脱泡,得到透明均一的电解质溶液(ionomer),溶液浓度依据实际需要来定。
步骤(3)、靛红芳烃共聚物阴离子交换膜的制备
将步骤(2)得到的浓度为3-15wt%电解质溶液铺膜,将膜浸入室温~80℃,1mnaoh或koh溶液进行离子交换2~24h,用室温去离子水浸泡24~48h、水洗至中性,干燥,得到靛红芳烃共聚物阴离子交换膜;其中,当靛红芳烃共聚物的结构通式中的r为烯烃、苯乙烯苄基、环氧基或丙烯酸酯时,在制膜时加入热引发剂或光引发剂得到交联型靛红芳烃共聚物阴离子膜;当r为二溴代烷时,在制膜时加入二元胺或三元胺进行化学交联得到交联型靛红芳烃共聚物阴离子膜。
所述的溴代烷r1-br,其中,r1是甲基或c1~c12的烷基。
上述的一种靛红芳烃共聚物电解质溶液(ionomer)在燃料电池中作为树脂粘结剂用于膜电极的制备。
上述的靛红芳烃共聚物阴离子交换膜在燃料电池、液流电池、电解、电渗析或分离膜领域的应用。
一种靛红芳烃共聚物、电解质溶液及其离子交换膜的合成路线如下:
一种靛红芳烃共聚物用于制备复合膜的方法,具体步骤如下:
步骤(1)、带铵阳离子的靛红芳烃共聚物的合成
将靛红芳烃共聚物在溶剂a中溶解得到5-20wt%溶液,加入碘甲烷或溴代烷r1-br,其中,碘甲烷或r1-br与靛红芳烃共聚物的摩尔比为(5~15):1,在30~100℃下反应20~100h;反应结束后,将反应液倒入沉淀剂中,过滤,水洗产物,得到带铵阳离子的靛红芳烃共聚物,其结构如通式(ⅱ)所示:
其中,0<n<1;x-是反离子,为br-或oh-离子;r1是甲基或c1~c12的烷基。
所述的溶剂a是n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、氯仿、二甲基亚砜、n-甲基吡咯烷酮中一种或两种以上混合。
所述的沉淀剂为甲醇、乙醇、水、石油醚、乙酸乙酯、丙酮、乙醚中一种或两种以上混合。
步骤(2)、带铵阳离子的靛红芳烃共聚物溶液的制备
将步骤(1)制备的带铵阳离子的靛红芳烃共聚物加入溶剂a中,搅拌溶解配制1~10wt%溶液,脱泡,得到透明均一的带铵阳离子的靛红芳烃共聚物溶液。
步骤(3)、复合膜的制备
将基膜室温浸泡在乙醇中3~24h,将基膜取出平铺,在表面均匀滴加少量溶剂a使基膜浸润,然后将步骤(2)制备的带铵阳离子的靛红芳烃共聚物溶液浇铸在基膜上自流平,将基膜放入干燥箱干燥至恒重;然后,将得到的基膜浸入室温~80℃,1mnaoh中2~24h离子交换,放在室温去离子水中24~48h,取出,真空干燥至恒重,得到靛红芳烃共聚物的复合膜。
所述的基膜为聚四氟乙烯或聚乙烯微孔膜,孔隙率>90%。
制备得到的靛红芳烃共聚物的复合膜应用于燃料电池、液流电池、电解、电渗析或分离膜。
与现有技术相比,本发明的有益效果:
按照本发明制备的带铵阳离子的靛红芳烃共聚物在25℃、n,n-二甲基乙酰胺中特性粘度为1.5~4.0dl/g。按照本发明制备的靛红芳烃共聚物的电解质溶液及其阴离子交换膜,离子传导率高,例如80℃时oh-离子传导率可达100ms/cm,湿膜及干膜均具有很强的韧性,机械性能优良,在80℃,1mnaoh溶液中浸泡30-40天,离子传导率下降较少,耐碱稳定性优良,在燃料电池、液流电池、电解及电渗析等离子膜相关领域有广泛应用。
附图说明
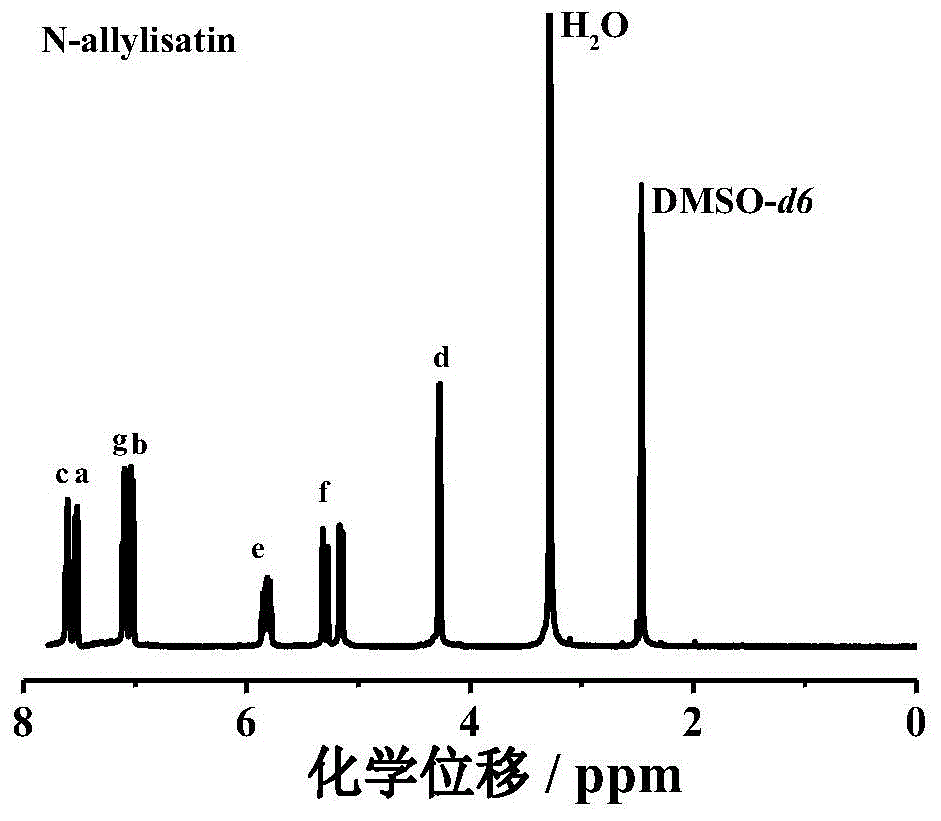
图1是实施例1中烯丙基靛红的1h-nmr谱图。
图2是实施例1中螺环铵盐靛红-哌啶酮-联苯共聚物的1h-nmr谱图。
图3是实施例8中己基甲基-哌啶盐靛红-甲基哌啶酮-联苯共聚物的1h-nmr谱图。
图4是实施例6中烯丙基靛红-甲基哌啶酮-三联苯共聚物的1h-nmr谱图。
具体实施方式
以下通过实施例进一步详细说明本发明靛红芳烃共聚物、制备方法及其应用,但不用于限制本发明的保护范围。
实施例所涉及的测试方法:
iec测试方法:取三份靛红芳烃共聚物0.2g左右,分别浸泡在100ml1mol/lnacl溶液24h。再用去离子水浸泡24h,真空烘箱干燥,称量记录。然后分别浸泡在25ml的0.5mnano3溶液中24h,并加入铬酸钾溶液指示剂,用0.1m的agno3溶液滴定,当出现砖红色沉淀并在30秒不变色即代表滴定完成,记录消耗的agno3溶液体积。将agno3溶液的浓度与体积的乘积除以干燥后膜的质量,即为iec。
耐碱稳定性:将膜放入80℃,1m的naoh溶液中浸泡,间隔一定时间将膜取出,用去离子水洗净到中性,测试室温导电率。
实施例1
将10mol靛红溶解在二甲基亚砜中,加入15mol无水k2co3室温搅拌溶解。加入12mol溴代丙烯,在60℃反应24h。将反应溶液倒入冰水中,用乙醚萃取,旋蒸除乙醚,产物用乙醇重结晶,得到烯丙基靛红。
将烯丙基靛红、4-哌啶酮盐酸盐加入反应器,加入联苯,加入二氯甲烷0℃搅拌溶解使单体浓度为20wt%。然后加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应20h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到烯丙基靛红-哌啶酮-联苯共聚物。其中烯丙基靛红与哌啶酮单体的摩尔比为1:4,烯丙基靛红和4-哌啶酮盐酸盐的总摩尔数与联苯的摩尔数之比为1:1,三氟甲磺酸与联苯的摩尔比为8:1,三氟乙酸与三氟甲磺酸体积比为1:8。
其中,烯丙基靛红-哌啶酮-联苯共聚物的哌啶结构n-h与稍过量的1,5-二溴戊烷,在催化剂作用下发生成环反应,生成螺环铵化的共聚物。具体反应如下:
将1.3mol上述共聚物溶解在n,n-二甲基甲酰胺配制5wt%溶液,滴加1.5mol1,5-二溴戊烷的dmac溶液,加入1.5mol的n,n-二异丙基乙胺作催化剂,在60℃反应36h。反应结束,将其倒入丙酮沉淀,过滤,用丙酮洗涤,水洗,真空干燥,得到螺环铵化的烯丙基靛红-哌啶酮-联苯共聚物。
取上述螺环铵化的共聚物溶于n-甲基吡咯烷酮中配制铸膜液。浇在洁净玻璃板上,干燥。把膜浸泡在80℃,1mnaoh中离子交换2h,再浸泡在去离子水中24h,用水洗涤,得到共聚物阴离子交换膜。
将上述螺环铵化共聚物溶于n-甲基吡咯烷酮中,配制10wt%的铸膜液。加入少量2,2-(1,2-乙二基双氧代)双乙硫醇作交联剂并加入光引发剂。将铸膜液浇在干净的玻璃板上,用365nm紫外灯照射进行交联。烘箱干燥,得到交联型螺环铵化共聚物阴离子膜。
螺环铵化共聚物的结构式如下:
图1是烯丙基靛红的1h-nmr谱图。化学位移在7.53和7.6,7.09和7.01ppm处为靛红苯环上质子的化学位移,化学位移在5.8、5.32和5.15ppm处为烯烃靛红的双键上质子的化学位移,化学位移在4.27ppm处为与靛红的n原子相连亚甲基上质子的化学位移。以上分析表明成功合成了烯丙基靛红。
图2是烯丙基靛红-哌啶酮-联苯共聚物和螺环铵化的聚烯丙基靛红-哌啶酮-联苯阴离子膜的1h-nmr谱图。从图2可见,化学位移在7.09-7.6ppm处的峰为苯环上质子峰,在8.9ppm处为哌啶上n-h的特征峰,在5.9、5.16和1.9ppm处为连接在靛红n原子上双键氢的特征峰,在3.06、2.69和2.18ppm处是哌啶环上亚甲基的质子峰。在哌啶酮上n原子成环后,其n-h峰消失,并出现1.76和1.56ppm处的化学位移峰,这是n螺环上质子的特征峰。由此表明上述共聚物和螺环铵化的共聚物阴离子膜已成功合成。所制得的阴离子膜为透明淡黄色,具有较好的柔韧性。
经实验测试:离子交换容量iec是1.03mmol/g,80℃时氢氧根离子传导率为53ms/cm,80℃时吸水率为45%,溶胀度为5.5%。在80℃,1m的naoh溶液中浸泡570h离子传导率保留率为90%。
实施例2
烯丙基靛红的合成:方法同实施例1,将10mol靛红溶解在二甲基亚砜中,加入15mol无水k2co3室温搅拌溶解。加入10mol溴代丙烯,在20℃反应50h。将反应溶液倒入冰水中,用乙醚萃取,旋蒸除乙醚,产物用乙醇重结晶,得到烯丙基靛红。
将烯丙基靛红、n-甲基-4-哌啶酮加入反应器,加入联苯。加入二氯甲烷在0℃搅拌溶解使单体浓度为20wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应14h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到烯丙基靛红-甲基哌啶酮-联苯共聚物。其中,烯丙基靛红与n-甲基-4-哌啶酮的摩尔比为2:3,烯丙基靛红和n-甲基-4-哌啶酮的总摩尔数与联苯的摩尔数之比为1:1,三氟甲磺酸与联苯的摩尔比为6:1,三氟乙酸与三氟甲磺酸体积比为1:14。
将2mol上述共聚物溶解在n,n-二甲基乙酰胺中溶解配制15wt%溶液,加入30mol碘甲烷在40℃反应24h。将聚合物溶液倒入丙酮析出,过滤,水洗,烘箱干燥,得到铵化的共聚物。取上述铵化的共聚物加入二甲基乙酰胺中,搅拌溶解得到澄清的电解质溶液(ionomer)。将其浇在洁净玻璃板上,60℃烘箱干燥,得到该铵化共聚物的阴离子交换膜。膜厚由溶液浓度来调整,一般铸膜液浓度为5-15wt%。
或者,取上述铵化的共聚物溶于二甲基乙酰胺中,配制8wt%的铸膜液。将其浇在洁净玻璃板上,加入光引发剂置于365nm紫外灯下照射30min,得到交联型铵化共聚物的阴离子交换膜。
铵化的共聚物的结构式如下:
80℃时氢氧根离子传导率100ms/cm,耐碱性在80℃,1mnaoh中1800h,导电率下降15%。
实施例3
己基靛红合成:用溴代己烷代替溴代丙烯,方法同实施例1,其中,将10mol靛红溶解在二甲基亚砜中,加入20mol无水k2co3室温搅拌溶解。加入30mol溴代己烷,在50℃反应10h。将反应溶液倒入冰水中,用乙醚萃取,旋蒸除乙醚,产物用乙醇重结晶,得到已基靛红。
将己基靛红、n-甲基-4-哌啶酮加入反应器,加入对三联苯。加入二氯甲烷0℃搅拌溶解使单体浓度为30wt%。加入三氟甲磺酸和三氟乙酸,在5℃搅拌反应48h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到己基靛红-甲基哌啶酮-三联苯共聚物。其中己基靛红与n-甲基-4-哌啶酮的摩尔比为1:4,己基靛红和n-甲基-4-哌啶酮的总摩尔数与对三联苯的摩尔数之比为1:1,三氟甲磺酸与对三联苯的摩尔比为12:1,三氟乙酸与三氟甲磺酸体积比为1:8。
将10mol上述共聚物溶解在n,n-二甲基甲酰胺中溶解配制10wt%溶液,加入50mol碘甲烷在30℃反应100h。将聚合物溶液倒入乙醇析出,过滤,水洗,烘箱干燥,得到铵化的共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜,其中,把膜浸泡在室温下,1mnaoh中离子交换24h,再浸泡在去离子水中48h,用水洗涤。
铵化的共聚物的结构式如下:
80℃时氢氧根离子传导率85ms/cm,耐碱性在80℃,1mnaoh中1700h,导电率下降15%。
实施例4
将靛红、n-甲基-4-哌啶酮加入反应器,加入联苯。加入二氯甲烷0℃搅拌溶解使单体浓度为35wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应20h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到靛红-甲基哌啶酮-联苯共聚物。其中,靛红与n-甲基-4-哌啶酮的摩尔比为15:85,靛红和n-甲基-4-哌啶酮的总摩尔数与联苯的摩尔数之比为1.1:1,三氟甲磺酸与联苯的摩尔比为10:1,三氟乙酸与三氟甲磺酸体积比为1:12。
将10mol上述共聚物溶解在二甲基乙酰胺中配制10wt%溶液,加入60mol碘甲烷在60℃反应48h。将聚合物溶液倒入丙酮析出,过滤,水洗,烘箱干燥,得到铵化靛红-甲基哌啶酮-联苯共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜,其中,把膜浸泡在80℃,1mnaoh中离子交换2h,再浸泡在去离子水中24h,用水洗涤。
图3是铵化的靛红-甲基哌啶酮-联苯共聚物的1h-nmr谱图。化学位移在10.84ppm为靛红中酰胺键的n-h出峰,在7.05-7.58ppm处特征峰为苯环上质子的化学位移,在3.15ppm处为哌啶n原子上甲基质子的特征峰,在2.84ppm处为哌啶环上亚甲基的质子的特征峰。说明成功合成了甲铵化的靛红-甲基哌啶酮-联苯共聚物。
铵化共聚物的结构式如下:
80℃时氢氧根离子传导率85ms/cm,耐碱性在80℃,1mnaoh中1700h,导电率下降15%.
实施例5
乙烯基苄基靛红合成,同实施例2。
将乙烯基苄基靛红、n-甲基-4-哌啶酮加入反应器,加入间三联苯。加入二氯甲烷0℃搅拌溶解使单体浓度为28wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到乙烯基苄基靛红-甲基哌啶酮-联苯共聚物。其中乙烯基苄基靛红与n-甲基-4-哌啶酮的摩尔比为3:7,乙烯基苄基靛红和n-甲基-4-哌啶酮的总摩尔数与间三联苯的摩尔数之比为1.05:1,三氟甲磺酸与间三联苯的摩尔比为6:1,三氟乙酸与三氟甲磺酸体积比为1:10。
将10mol上述共聚物溶解在n,n-二甲基乙酰胺中溶解配制3wt%溶液,加入60mol溴己烷在50℃反应36h。将聚合物溶液倒入甲醇析出,过滤,水洗,烘箱干燥,得到铵化共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜。
交联型铵化离子膜的制备,同实施例2。
铵化共聚物的结构式如下:
80℃时氢氧根离子传导率80ms/cm,耐碱性在80℃,1mnaoh中2000h,导电率下降15%。
实施例6
烯丙基靛红合成,同实施例1。
将烯丙基靛红、n-甲基-4-哌啶酮加入反应器,加入对三联苯。加入二氯甲烷0℃搅拌溶解使单体浓度为30wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应22h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到烯丙基靛红-甲基哌啶酮-三联苯共聚物。其中烯丙基靛红与n-甲基-4-哌啶酮的摩尔比为1:9,烯丙基靛红和n-甲基-4-哌啶酮的总摩尔数与对三联苯的摩尔数之比为1.1:1,三氟甲磺酸与对三联苯的摩尔比为9:1,三氟乙酸与三氟甲磺酸体积比为1:10。
将10mol上述共聚物溶解在n,n-二甲基乙酰胺中溶解配制8wt%溶液,加入100mol碘甲烷在70℃反应48h。将聚合物溶液倒入乙醚析出,过滤,水洗,烘箱干燥,得到铵化共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜。
交联型铵化离子膜的制备,同实施例2。
铵化共聚物的结构式如下:
80℃时氢氧根离子传导率80ms/cm,耐碱性在80℃,1mnaoh中2000h,导电率下降15%。
图4是实施例6烯丙基靛红-甲基哌啶酮-三联苯共聚物的1h-nmr图。
实施例7
己基靛红的合成:用溴代己烷代替溴代丙烯,其它反应同实施例1。
将己基靛红、n-甲基-4-哌啶酮加入反应器,加入联苯。加入二氯甲烷0℃搅拌溶解使单体浓度为15wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应30h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到己基靛红-甲基哌啶酮-联苯共聚物。其中己基靛红与n-甲基-4-哌啶酮的摩尔比为1:4,己基靛红和n-甲基-4-哌啶酮的总摩尔数与联苯的摩尔数之比为1:1,三氟甲磺酸与联苯的摩尔比为14:1,三氟乙酸与三氟甲磺酸体积比为1:10。
将1mol上述共聚物溶解在n,n-二甲基乙酰胺中溶解配制8%溶液,加入10mol碘甲烷在30℃反应24h。将聚合物溶液倒入甲醇析出,过滤,水洗,烘箱干燥,得到铵化共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜。
铵化共聚物的结构式如下:
80℃时氢氧根离子传导率85ms/cm,耐碱性在80℃,1mnaoh中1600h,导电率下降15%。
实施例8
溴代己基n-甲基哌啶盐由n-甲基哌啶和过量的二溴己烷经门秀金反应合成。
n-甲基己基哌啶盐靛红的合成:用溴代己基n-甲基哌啶盐代替溴代丙烯,其它反应同实施例1。
将n-甲基己基哌啶盐靛红、n-甲基-4-哌啶酮加入反应器,加入联苯。加入二氯甲烷0℃搅拌溶解使单体浓度为20wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应38h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到己基甲基哌啶盐靛红-甲基哌啶酮-联苯共聚物。其中n-甲基己基哌啶盐靛红与n-甲基-4-哌啶酮的摩尔比为2:3,n-甲基己基哌啶盐靛红和n-甲基-4-哌啶酮的总摩尔数与联苯的摩尔数之比为1:1,三氟甲磺酸与联苯的摩尔比为7:1,三氟乙酸与三氟甲磺酸体积比为1:8。
将3mol上述共聚物溶解在n,n-二甲基乙酰胺中溶解配制8%溶液,加入20mol碘己烷在60℃反应24h。将聚合物溶液倒入甲醇析出,过滤,水洗,烘箱干燥,得到铵化共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜。
铵化共聚物的结构式如下:
80℃时氢氧根离子传导率110ms/cm,耐碱性在80℃,1mnaoh中1800h,导电率下降15%。
实施例9
乙烯基苄基靛红的合成同实施例2。
将乙烯基苄基靛红、n-甲基-4-哌啶酮加入反应器,加入双酚芴。加入二氯甲烷0℃搅拌溶解使单体浓度为35wt%。加入三氟甲磺酸和三氟乙酸,在0℃搅拌反应20h,当体系变得粘稠将聚合产物倒入大量冰水,用1mnahco3除去过量的酸,水洗产物,过滤,真空干燥,得到乙烯基苄基靛红-甲基哌啶酮-双酚芴共聚物。其中乙烯基苄基靛红与n-甲基-4-哌啶酮的摩尔比为1:4,乙烯基苄基靛红和n-甲基-4-哌啶酮的总摩尔数与双酚芴的摩尔数之比为1:1,三氟甲磺酸与对双酚芴的摩尔比为8:1,三氟乙酸与三氟甲磺酸体积比为1:10。
将10mol上述共聚物溶解在二甲基亚砜中溶解配制10wt%溶液,加入60mol碘甲烷在50℃反应48h。将聚合物溶液倒入乙醚析出,过滤,水洗,烘箱干燥,得到铵化共聚物。
采用实施例2方法,制得该铵化共聚物的电解质溶液及阴离子交换膜。
交联型铵化离子膜的制备,同实施例2。
铵化共聚物的结构式如下:
80℃时氢氧根离子传导率75ms/cm,耐碱性在80℃,1mnaoh中2000h,导电率下降15%。
实施例10
将实施例2制备的铵化的烯丙基靛红-甲基哌啶酮-联苯共聚物加入二甲基亚砜中,搅拌溶解配制3wt%溶液,脱泡,得到透明均一的共聚物溶液。
室温时,将2.0×3.0cm2的聚四氟乙烯ptfe微孔膜浸泡乙醇中10h。将ptfe膜取出平铺,用滴管均匀滴加二甲基亚砜,用滤纸把膜表面的液体吸干。将上述共聚物溶液滴加在ptfe膜表面自流平,然后将膜放入烘箱60℃干燥至恒重。将得到的膜浸入80℃,1mnaoh中2h离子交换,然后放在室温去离子水中24h,干燥至恒重,得到复合膜。
80℃时氢氧根离子传导率60ms/cm,耐碱性在80℃,1mnaoh中2300h,导电率下降12%。
实施例11
将实施例6制备的铵化的烯丙基靛红-甲基哌啶酮-三联苯加入二甲基乙酰胺dmac中,搅拌溶解配制1wt%溶液,脱泡,得到透明均一的共聚物溶液。
室温时,将2.0×3.0cm2的聚乙烯微孔膜浸泡乙醇中3h。将聚乙烯微孔膜取出平铺,用滴管均匀滴加几滴dmac,用滤纸把膜表面的液体吸干。将上述铵化的共聚物溶液滴加在ptfe膜表面自流平,然后将膜放入烘箱60℃干燥至恒重。将得到的膜浸入室温下,1mnaoh中24h离子交换,然后放在室温去离子水中48h,取出,干燥至恒重,得到复合膜。
实施例12
将实施例8制备的铵化的己基甲基哌啶盐靛红-甲基哌啶酮-联苯共聚物加入dmac中,搅拌溶解配制10wt%溶液,脱泡,得到透明均一的共聚物溶液。
室温时,将2.0×3.0cm2的聚四氟乙烯ptfe微孔膜浸泡乙醇中24h。将ptfe膜取出平铺,用滴管均匀滴加dmac溶剂,用滤纸把膜表面的液体吸干。将上述共聚物溶液滴加在ptfe膜表面自流平,然后将膜放入烘箱60℃干燥至恒重。将得到的膜浸入80℃,1mnaoh中6h离子交换,然后放在室温去离子水中30h,干燥至恒重,得到复合膜。
- 还没有人留言评论。精彩留言会获得点赞!