一种咔唑类热激活延迟荧光材料及制备方法与流程
本发明属于有机电致发光显示技术领域,涉及一种咔唑类热激活延迟荧光材料的制备。
背景技术:
近年来,在有机发光二极管(oleds)领域,热激活延迟荧光(tadf)发射器由于能够通过有效的系统间反向交叉(risc)过程实现近100%的内量子效率(iqe)而受到世界范围内的广泛关注。与磷光配合物相比,tadf材料具有明显的资源优势和较低的成本。
自从具有热活化延迟荧光现象(tadf)的有机材料被成功合成,tadf材料便引起了国内外众多科研机构和公司极大的关注。tadf材料的单线态和三线态能级差很小,使得他的三线态能通过反向系间窜跃上转化到单线态,最后发射荧光。tadf材料具有高内量子效率,原材料易获取,制备成本低廉等优点,tadf材料被认为是能极大推动有机发光二极管(oleds)产业化的第三代发光材料。tadf材料在oleds器件中的应用研究成为了研究热点之一。高效率长寿命的白光oleds器件是实现oleds产业化的必要保证,因此如何制备基于tadf材料的高效白光oleds成为了一个研究热点。这是目前基于tadf材料发光的外量子效率最高的oled。
因此,tadf化合物被认为是下一代oled材料。自从tadf的开创性工作以来,目前已经开发出大量的tadf发射器。开发高效的tadf材料,两个关键的要求,即,足够的risc流程和高φpl,应该满足。因此合成新型tadf分子的设计仍然是一个巨大的挑战。
总而言之,研发新型tadf材料应用于目前oled显示技术中具有高色纯度和高发光效率的要求,新型配体的开发应用是解决这一问题的有效途径。
技术实现要素:
本发明的目的是合成一种新型咔唑类热激活延迟荧光材料的制备。
一种新型咔唑类热激活延迟荧光材料的制备方法,为了解决上述问题,本发明所采用的技术方案为,
一种新型咔唑类热激活延迟荧光材料,该荧光材料的结构为如式i-1~4所示,
上述一种新型咔唑类热激活延迟荧光材料的制备方法,按照下述步骤进行:
(a)将咔唑,碱性试剂与2,3,5,6-四氟-4-碘苯氰溶于有机溶剂中,在回流条件下进行反应得到结构式i-3所示的化合物,
(b)将咔唑,碱性试剂与2',4',6'-三氟-[1,1':3',1”-三联苯]-5'-甲腈溶于有机溶剂中,在回流条件下进行反应得到结构式i-4所示的化合物,
(c)将步骤(a)所得的的结构式i-3,碱性试剂与4,4'-二苯基咔唑溶于有机溶剂中,在回流条件下进行反应得到结构式i-1所示的化合物,
(d)将步骤(b)所得的构式所示的i-4,碱性试剂与4,4'-二苯基咔唑溶于有机溶剂中,在回流条件下进行反应得到结构式i-2所示的化合物,
其中步骤a,b,c,与d中,加入碱性试剂为氢化钠或者叔丁醇钾,其用量与式i-1,2,3,5,6-四氟-4-碘苯氰的摩尔比为2.2:1,优选为2.5:1,进一步优选为3.0:1;所使用溶剂四氢呋喃与式i-1,咔唑摩尔比为4~5:1,优选为4.0~4.5:1,进一步优选为4.5~5:1。
其中步骤a和b中,加入咔唑的其用量与式i-3与i-4所示的2,3,5,6-四氟-4-碘苯氰的摩尔比为1.1~1:1,优选为1.2~1.3:1,进一步优选为1.3~1.4:1;所使用溶剂二甲基甲酰胺或四氢呋喃与式i-3与i-4所示的2,3,5,6-四氟-4-碘苯氰的摩尔比为4~5:1,优选为4.0~4.5:1,进一步优选为4.5~5:1。
其中步骤c和d中,所加入i-3与i-4的其用量与式i-1与i-2所示的与4,4'-二苯基咔唑的摩尔比为2.2~2:1,优选为2.4~2.3:1,进一步优选为2.5~2.4:1;所使用溶剂四氢呋喃与式i-1与i-2所示的4,4'-二苯基咔唑的摩尔比为4~5:1,优选为4.0~4.5:1,进一步优选为4.5~5:1。
步骤a所述i-3过程为将咔唑与2,3,5,6-四氟-4-碘苯氰溶于有机溶液中,降温至0~5℃后,向其中加入氢化钠,整个过程中保持反应温度为0~5℃,加入完毕后升温至90~100℃,继续反应4~6h,所得的反应溶液经减压蒸馏除去溶剂后,得到式i-3所示的化合物。
步骤c所述i-1过程为将i-3与4,4'-二苯基咔唑溶于有机溶液中,降温至5~10℃后,向其中加入氢化钠,整个过程中保持反应温度为0~5℃,加入完毕后升温至60~70℃,继续反应14~16h,所得的反应溶液经减压蒸馏除去溶剂后,得到式i-1所示的化合物。
所述i-4与i-2过程可参考上述i-3与i-1所示方法。
效果预测:该结构为新结构新物质,而且方法原料廉价易得,反应步骤简单,无刻薄危险反应条件,产品易提纯,适合于国内解决该领域空白技术。
附图说明
图1为实施例1中步骤1制备化合物的氢谱图;
图2为实施例1中步骤1制备化合物的氟谱图;
图3为实施例1中步骤2制备化合物的氢谱图;
图4为实施例2中步骤3制备化合物的氢谱图;
图5为实施例2中步骤3制备化合物的氟谱图;
图6为实施例2中步骤4制备化合物的氢谱图;
图7为实施例2中步骤4制备化合物的氟谱图。
具体实施方式
为了加深对本发明的理解,下面将结合实施例和附图对本发明作进一步详述,该实施例仅用于解释本发明,并不构成对本发明保护范围的限定。所述方法如无特别说明均为常规方法、所用原材料如无特别说明均能从公开商业途径获得。
本发明提供的式i-1~4所示化合物,其制备方法可按如下反应式制备而得:
实施例1化合物i-1和i-3的制备方法
1)称取2.0g咔唑,3.6g2,3,5,6-四氟-4-碘苯氰和1.6g叔丁醇钾溶于50ml二甲基甲酰胺后,加入到三口瓶之中,温度升至100度,反应时间为4小时,待反应完毕,温度降至室温,过滤,减压蒸馏有机溶剂,加入乙酸乙酯20毫升所生成的固体进行过滤洗涤得到i-3化合物3.6g,收率为89%。
检测参数如下:
1h-nmr:6.65-6.48(m,3h),6.80-6.73(m,2h),7.30-7.23(m,2h),7.41-7.31(m,4h),7.52-7.45(m,6h),7.60-7.58(m,2h),7.66-7.64(m,2h),7.73-7.71(m,2h),8.05-8.03(m,1h),18.10(s,1h),8.31(s,1h)。
19f-nmr:-130.60,-138.25。
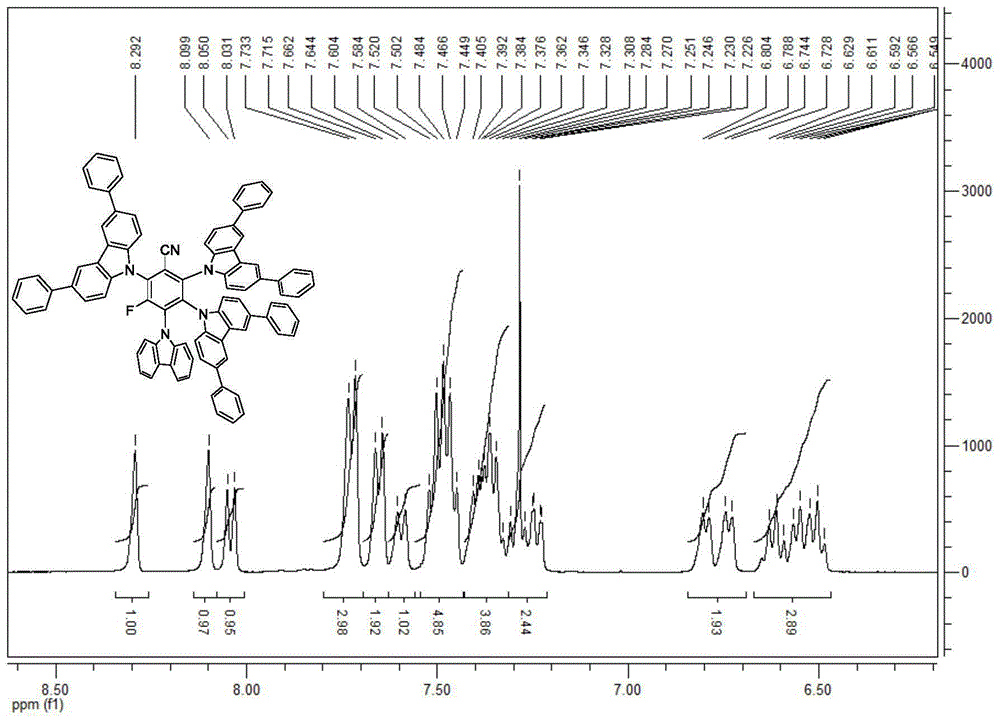
2)称取3.0gi-3和6.2g4,4'-二苯基咔唑溶于80ml四氢呋喃后,加入到三口瓶之中,缓慢加入0.8g(60%)氢化钠后,温度升至65度反应20小时,待反应完毕,温度降至室温,待反应完毕减压蒸馏有机溶剂,加入冰水100毫升,所生成的固体过滤,得到的固体加入甲醇100毫升,过滤,得到的固体再次加入二氯甲烷100毫升,过滤,干燥得到i-1化合物1.9g,收率为17%。
检测参数如下:
1h-nmr:7.02-6.98(m,2h),7.12-7.06(m,2h),7.29-7.18(m,4h),7.42-7.35(m,22h),7.62-7.55(m,10h),7.85-7.77(m,8h),7.98-7.94(m,4h),8.58-8.57(m,4h)。
19f-nmr:-113.35。
详细见图1(实施例1中步骤1制备化合物的氢谱图)、图2(实施例1中步骤1制备化合物的氟谱图)和图3(实施例1中步骤2制备化合物的氢谱图)。
实施例2化合物i-2和i-4的制备方法
1)称取2.0g2',4',6'-三氟-[1,1':3',1”-三联苯]-5'-甲腈和1.2g咔唑溶于80ml二甲基甲酰后,加入到三口瓶之中,缓慢加入0.3g(60%)氢化钠后,温度升至100度反应14小时,待反应完毕,温度降至室温,待反应完毕减压蒸馏有机溶剂,加入冰水100毫升,所生成的固体过滤,得到的固体加入甲醇100毫升,过滤,得到的固体再次加入乙酸乙酯100毫升,过滤,干燥得到i-4化合物2.1g,收率为72%。
检测参数如下:
1h-nmr:7.18-7.13(m,1h),7.42-7.40(m,1h),7.53-7.49(m,1h),8.18-8.15(d,j=8.17hz,1h)。
19f-nmr:-113.35。
2)称取2.0gi-4和3.0g4,4'-二苯基咔唑溶于80ml四氢呋喃后,加入到三口瓶之中,缓慢加入0.3g(60%)氢化钠后,温度升至65度反应18小时,待反应完毕,温度降至室温,待反应完毕减压蒸馏有机溶剂,加入冰水100毫升,所生成的固体过滤,得到的固体加入甲醇100毫升,过滤,得到的固体再次加入二氯甲烷100毫升,过滤,干燥得到i-2化合物1.0g,收率为21%。
检测参数如下:
1h-nmr:7.02-6.96(m,2h),7.18-7.16(d,j=7.60hz,1h),7.28-7.25(m,1h),7.43-7.39(m,1h),7.65-7.58(m,2h),8.15-8.03(d,j=8.03hz,1h)。
19f-nmr:-101.79,-104.15,-105.09。
详细见图4(实施例2中步骤3制备化合物的氢谱图)、图5(实施例2中步骤3制备化合物的氟谱图)、图6(实施例2中步骤4制备化合物的氢谱图)和图7(实施例2中步骤4制备化合物的氟谱图)。
- 还没有人留言评论。精彩留言会获得点赞!