一种二苯基吡嗪衍生物的合成方法
本发明属于有机合成领域,具体涉及一种二苯基吡嗪衍生物及其合成方法。
背景技术:
肺动脉高压(pulmonaryarterialhypertension,pah)是一种以肺动脉压力增高,伴或不伴小肺动脉病变为特征的恶性肺血管疾病,往往引起右心功能衰竭甚至死亡。pah可以是一种独立的疾病,也可是并发症,还可以是综合征。美国nih报道pah的自然中位生存期仅2.8年,有心血管系统的“恶性肿瘤”之称。进年来,随着靶向药物上市,诊治手段的提高,pah患者的生存率显著改善,但仍然不能治愈,五年生存率仍仅60%。
前列环素(pgi2)及其类似物可用于治疗各种血管疾病,但其半衰期太短,无法广泛应用于临床。为了克服这个缺点,由瑞士生物科技企业actelion和日本新药株式会社联合开发了一种新的二苯基吡嗪衍生物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺,是一种选择性口服ip环前列腺素受体激动剂。2015年12月22日,美国食品药品监督管理局(fda)批准actelion的肺动脉高压新药
目前,文献报道的2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺的合成路线有两种,均采用了5-氯-2,3-二苯基吡嗪作为起始物料,通过一步或两步制备了4-[(5,6-二苯基-2-吡嗪基)(1-甲基乙基l)氨基]-1-丁醇(中间体ⅱ)。
路线1
路线2
路线1通过中间体ⅱ经缩合、水解制备了2-{4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}乙酸(ns-303),再与甲基磺酰胺酰胺缩合反应得到目标产物。路线2通过中间体ⅱ直接与2-氯-n-(甲磺酰)乙酰胺缩合制得目标产物。两条路线中起始物料5-氯-2,3-二苯基吡嗪不稳定,不易长期存储,且在制备中间体ⅱ时都采用了超高温反应170℃-200℃,存在一定的安全隐患。
技术实现要素:
本发明的目的在于提供一种二苯基吡嗪衍生物的合成方法。
为达到上述目的,本发明的技术方案是:
一种二苯基吡嗪衍生物的合成方法,以苯偶酰为原料,经五步反应得到目标化合物,包括:
(1)将苯偶酰与甘氨酰在碱性条件下加热至70℃发生反应,制备5,6-二苯基-2-羟基吡嗪(ns-300)的步骤,
(2)在无水溶剂中,将5,6-二苯基-2-羟基吡嗪和三溴氧磷在碱催化剂作用下,发生反应制备5-溴-2,3-二苯基吡嗪(ns-301)的步骤,
(3)在二氧六环溶剂中,将5-溴-2,3-二苯基吡嗪与异丙胺在碱性条件下,发生反应制备n-异丙基-5,6-二苯基吡嗪-2-胺(ns-302)的步骤,
(4)在乙腈溶剂中,将2-(4-氯丁氧基)乙酸与n-异丙基-5,6-二苯基吡嗪-2-胺在碱性条件下,发生反应制备2-{4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}乙酸(ns-303)的步骤,
(5)在thf溶剂中,将2-{4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}乙酸与甲基磺酰胺,在催化剂n,n’-羰基二咪唑(cdi)和1,8-二氮杂双环[5,4,0]十一碳-7-烯(dbu)共同作用下,发生反应制备目标产物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺(ns-304)的步骤,
较佳的,步骤(1)中,所述的碱性条件是指在氢氧化钠或氢氧化钾存在下;苯偶酰与甘氨酰的摩尔比为1:1.1。
较佳的,步骤(2)中,无水溶剂包括乙腈、甲苯、二甲苯、氯苯、四氢呋喃中任意一种;反应温度为回流温度下;碱催化剂为三乙胺、吡啶中任意一种;5,6-二苯基-2-羟基吡嗪和三溴氧磷的摩尔比为1:3.0。
较佳的,步骤(3)中,所述的碱性条件是指在碳酸钾、氢氧化钠、氢氧化钾中任意一种存在下;5-溴-2,3-二苯基吡嗪与异丙胺的摩尔比为1:1.5。
较佳的,步骤(4)中,所述的碱性条件是指在叔丁醇钾、叔丁醇钠,钠氢中任意一种存在下;以摩尔比计,n-异丙基-5,6-二苯基吡嗪-2-胺:溴乙酸:4-氯丁醇=1:3.5:2.5。
较佳的,步骤(5)中,反应温度为60℃-65℃温度下;2-{4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}乙酸和甲基磺酰胺的摩尔比为:1:1.0-1:2.0,优选1:1.5;2-{4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}乙酸与催化剂cdi和dbu的摩尔比为:1:1.2:1.5。
本发明与现有技术相比,其优点有:
本发明以苯偶酰和甘氨酰胺为原料,化学性质更稳定。该合成路线设计合理,反应条件温和,不存在高温反应,安全性更好、可行性更高,具有广阔的应用前景。本发明得到高纯度的目标产物,有利于后续生产高品质的药品。
附图说明
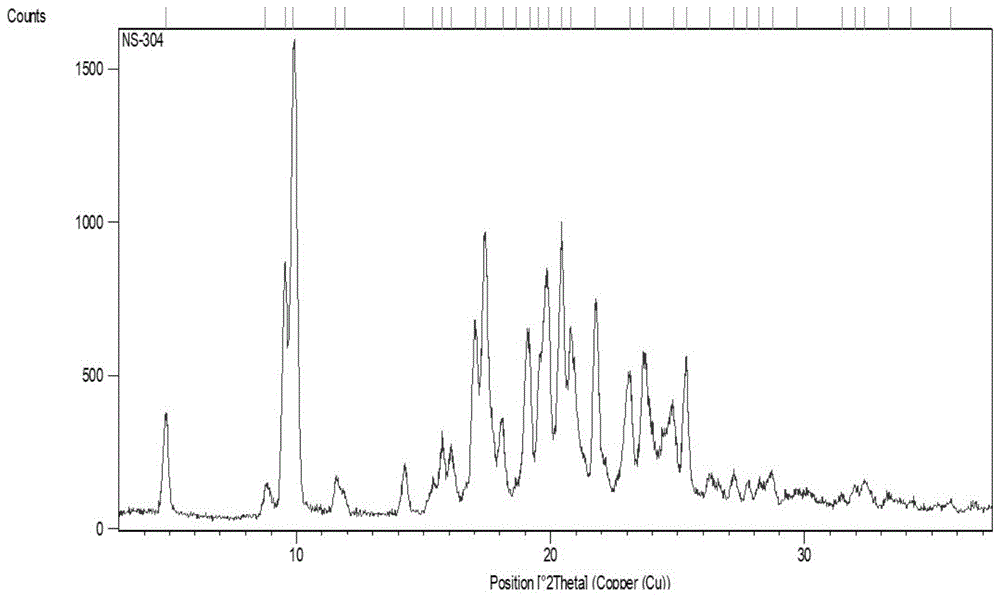
图1是目标产物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺的x射线衍射图。
图2是目标产物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺的1h-nmr图。
图3是目标产物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺的13c-nmr图。
图4是目标产物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺的esi-ms图。
具体实施方式
为了更好地理解本发明内容,以下结合实施例和附图详细说明了本发明,但本发明不仅局限于此。
本发明为克服现有合成路线的缺点,设计了以常规原料苯偶酰为起始物料的合成路线,经亲核加成、卤代、亲核取代、缩合、酰胺缩合得到目标化合物2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺。路线如下:
实施例所用原料和试剂均为市售产品。实施例中所述“%”均为重量百分比(wt%.),特别说明的除外。
实施例1
5,6-二苯基-2-羟基吡嗪(化合物ns-300)的合成
反应步骤:
向配有冷凝管的1000ml三口烧瓶中加入甲醇(350ml),加入苯偶酰(35.02g)与甘氨酰胺盐酸盐(19.75g,1.1eq),冰水浴冷却10℃,缓慢滴加12n的氢氧化钠溶液(30ml),控温≤30℃,滴加完毕,转油浴加热,升温70℃回流反应12h,hplc检测原料消失,停止加热,开启冷却降温至10℃,向体系中加入冰醋酸调节ph至6-7,复测ph不变。有大量淡黄色固体析出,抽滤,得亮黄色固体,70℃鼓风干燥,不经提纯直接投入下一步。
实施例2
5-溴-2,3-二苯基吡嗪(化合物ns-301)的合成
反应步骤:
向配有冷凝管的500ml三口烧瓶中加入乙腈(350ml),搅拌加入ns-300(30.00g),加入三溴氧磷(103.92g,3.0eq),搅拌升温至70℃,反应30h,hplc检测显示反应完全,将反应液缓慢加入850ml水中,淬灭反应,析出大量浅紫色固体,抽滤,滤饼用naoh稀溶液打浆,调节至中性,抽滤得固体33.46g。产率约89%。esi-ms:m/z=313.0(m+h)+。1hnmr(500mhz,dmso-d6)δ8.84(s,1h),7.43-7.30(m,10h)。
实施例3
n-异丙基-5,6-二苯基吡嗪-2-胺(化合物ns-302)的合成
反应步骤:
向500ml三口烧瓶中依次加入二氧六环(250ml)、ns-301(30.03g)、异丙胺(8.62g,1.5eq)、ki(4.80g,0.3eq)和碳酸钾(41.14g,2.0eq),搅拌均匀,加热到40-45℃反应12h,tlc监测反应完全。将反应混合物冷却至10℃左右,加水(100ml)淬灭反应并用二氯甲烷萃取。合并有机层用饱和食盐水洗并经无水硫酸钠干燥。蒸馏出溶剂,得到固体20.41g。收率约73%。esi-ms:m/z=290.4(m+h)+。
实施例4
2-{4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}乙酸(化合物ns-303)的合成
反应步骤:
250ml三口烧瓶中加入乙腈(100ml)、4-氯丁醇(13.82g)、叔丁醇钾(24.27g)搅拌均匀,冰水浴降温至10℃。缓慢滴加溴乙酸(25.06g),滴加完成转油浴加热搅拌反应6h,过滤得2-(4-氯丁氧基)乙酸的乙腈溶液。
250ml三口烧瓶中加入乙腈(60ml,4v)、ns-302(15.00g)和叔丁醇钾(16.32g)搅拌混合均匀,水浴10℃。控温低于10℃,将2-(4-氯丁氧基)乙酸的乙腈溶液缓慢滴加入反应体系。加热反应并在相同温度下搅拌14h,tlc监控反应结束。过滤出不溶物,分溜出一半乙腈后将反应混合物冷却至室温,液体用水稀释溶清。缓慢滴加稀盐酸至淡黄色固体全部析出,调节体系ph=2,过滤,得固体直接用乙醇重结晶,得黄色粉末状固体14.09g。收率约62%。其结构表征见图1-4。esi-ms:m/z=420.4(m+h)+。1hnmr(500mhz,dmso-d6)δ8.00(s,1h),7.33-7.17(m,10h),4.66(p,j=6.8hz,1h),3.99(s,2h),3.50(t,j=6.0hz,2h),3.39(t,j=7.6hz,2h),1.62(dh,j=21.5,8.2hz,4h),1.20(d,j=6.7hz,6h)。
实施例5
2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺(化合物ns-304)的合成
反应步骤:
将ns-303(10.00g)溶于thf(200ml)中,加入cdi(5.71g,1.5eq),升温至60℃-65℃反应1h,开始降至0-5℃,加入dbu(4.82ml,1.2eq),保持温度稳定,加入甲基磺酰胺(3.44g,1.5eq),60℃-65℃反应12h,tlc跟踪至产物点不再变化。加水(50ml)稀释反应液,再加入甲基叔丁基醚(50ml),搅拌均匀后静置分液,有机相用饱和食盐水萃洗,分液,无水硫酸钠干燥有机相,过滤出硫酸钠,浓缩有机相至无液体滴出,得油状物粗品13.51g。
实施例6
2-[4-[n’-(5,6-二苯基吡嗪-2-基)-n’-异丙基氨基]丁氧基}-n-(甲磺酰基)乙酰胺(化合物ns-304)的精制
ns-304粗品10.13g用100ml乙醇进行精制,加热回流溶清固体,冷凝分出60ml乙醇,溶液仍是澄清,梯度降温(10℃/h降温3h,后改为5℃/h降温至10℃左右)有晶体析出。抽滤得固体。重复以上精制步骤两次得固体5.97g,熔点140.7℃,纯度99.79%(hplc,面积归一化法),产率约59%。其结构表征见图1~4。esi-ms:m/z=497.3(m+h)+。1hnmr(500mhz,dmso-d6)δ8.04(s,1h),7.33-7.28(m,2h),7.28-7.24(m,2h),7.24-7.19(m,6h),4.66(p,j=6.8hz,1h),4.01(s,2h),3.50(t,j=6.0hz,2h),3.39(t,j=7.6hz,2h),3.21(s,3h),1.62(dh,j=21.5,8.2hz,4h),1.18(d,j=6.7hz,6h).13cnmr(500mhz,dmso-d6)δ19.90,25.55,26.57,41.18,41.78,45.77,68.97,70.66,126.78,127.49,127.79,127.89,128.00,129.03,129.37,138.04,139.29,139.45,148.01,151.14,169.95。
实施例7
条件同实施例4,调节溶液的ph值在3左右,收率58%。
实施例8
条件同实施例6,降温速率改为10℃/h降温至10℃,纯度99.56%,收率43%。
- 还没有人留言评论。精彩留言会获得点赞!