一种多取代二氢吡咯化合物的制备方法
本发明属于有机合成技术领域,具体涉及一类多取代二氢吡咯衍生物的制备方法。
背景技术:
吡咯类化合物是一类重要的五元含氮杂环化合物,该类杂环是许多药物和功能材料的核心结构单元,对于其生物活性和材料功能的发挥具有重要作用,在医药、农药、生物化学、天然产物合成、药物合成和材料化学等领域有广泛的应用。其中二氢吡咯类化合物和吡咯不仅可以相互转化,而且可以发生取代反应、氧化反应、蒂尔斯-阿德尔反应、环加成反应、偶联反应等多种化学反应,是合成吡咯以及其他杂环化合物等的重要中间体,在有机合成中有着广泛的应用。因此二氢吡咯类化合物的合成方法研究一直被人们所关注。
近年来,自由基环化和过渡金属催化合成二氢吡咯类化合物的方法取得了长足发展。举例如下:
(1)zard组,他们在1995年利用n-bu3snh/aibn体系将肟醚1转化为亚胺自由基,后者环化,生成二氢吡咯类化合物。
(2)weinreb组,在1999年也报道了类似γ,δ-不饱和肟的碳酸酯在锡氢条件下的自由基环化反应。
(3)narasaka组,2002年报道了cu催化下γ,δ-不饱和肟酯5的环化反应。在libr存在下,得到溴代二氢吡咯产物。
(4)苏建华课题组,在2015报道了苯基肟酯化合物在醋酸钯、膦配体p(4-fc6h4)3和lix条件下生成卤代二氢吡咯化合物。反应成功实现了对末端cl,br,i的官能团化。
以上几种方法的缺点是环化前体大多需事先制备分离,反应条件苛刻,原料制备复杂并且通常需要昂贵的过渡金属催化剂及复杂的配体,无法实现工业化应用。
技术实现要素:
本发明所要解决的技术问题在于针对上述现有技术的不足,提供一种多取代二氢吡咯化合物的制备方法。
为解决上述技术问题,本发明采用的技术方案是:在反应器中加入1,3-二噻烷类化合物i,取代亚胺ii,然后在氩气保护下;另将碱溶解在四氢呋喃中,然后将碱溶液缓慢的滴加到反应器中;最后在室温的条件下搅拌反应0.5~2小时(具体反应时间以薄层层析监测反应完全为准),对反应完全后的反应体系直接柱层析分离纯化,得到多取代二氢吡咯化合物iii,反应方程式如下:
方程式中:r1和r2为、苯基、取代苯基中的一种。
上述的一种多取代二氢吡咯化合物的制备方法,优选的溶剂为四氢呋喃。
上述的一种多取代二氢吡咯化合物的制备方法,优选的1,3-二噻烷化合物、亚胺和碱的用量的摩尔比为1:1.5:2。
上述的一种多取代二氢吡咯化合物的制备方法,优选的碱为叔丁醇钾。
本发明与现有技术相比具有以下优点:
1、本发明具有操作安全、环境友好、反应条件温和和收率高的优点。
2、本发明所用原料和碱廉价易得,环保压力小。
3、本发明符合绿色合成的要求,具有较大的实际应用价值。
下面通过实施例,对本发明技术方案做进一步的详细说明。
附图说明
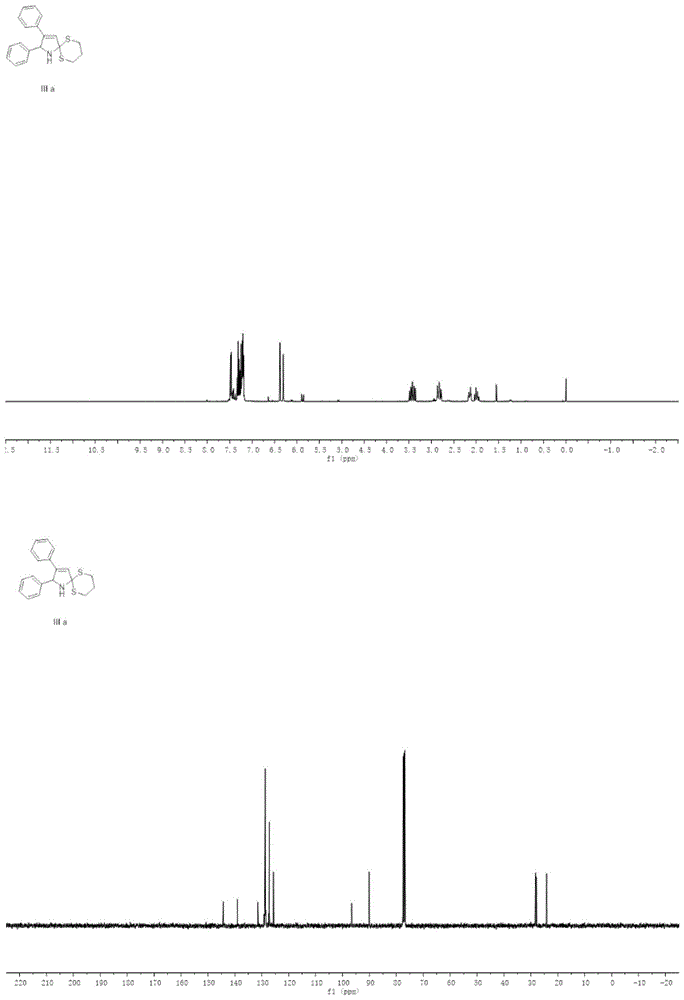
图1iiia的氢谱、碳谱数据
图2iiib的氢谱、碳谱数据
图3iiic的氢谱、碳谱数据
图4iiid的氢谱、碳谱数据
图5iiie的氢谱、碳谱数据
图6iiif的氢谱、碳谱数据
具体实施方式
实施例1
在10毫升圆底烧瓶中依次加入化合物ia(0.1mmol)、iia(0.15mmol)在氩气保护下;然后将叔丁醇钾(0.20mmol)溶解在无水的四氢呋喃中,并将其缓慢滴加如反应器中;,直至反应完全;加入5ml水,用10ml乙酸乙酯分三次萃取,合并乙酸乙酯溶液,用无水硫酸钠干燥,减压蒸出乙酸乙酯,剩余物用石油醚/乙酸乙酯=60/1的流动相经硅胶柱层析分离纯化,得到多取代吡咯化合物iiia,产率72%。
1hnmr(400mhz,cdcl3)δ7.49–7.47(m,2h),7.34–7.27(m,3h),7.26–7.19(m,6h),6.38(d,j=2.1hz,1h),6.31(d,j=2.1hz,1h),3.50–3.35(m,2h),2.87–2.78(m,2h),2.18–2.11(m,1h),2.04–1.96(m,1h);13cnmr(101mhz,cdcl3)δ144.2,139.1,131.4,128.8,128.7,128.7,128.6,127.1,125.6,96.6,90.0,77.4,77.1,76.8,28.2,27.9,24.1;
实施例2
在10毫升圆底烧瓶中依次加入化合物ia(0.1mmol)、iib(0.15mmol)在氩气保护下;然后将叔丁醇钾(0.20mmol)溶解在无水的四氢呋喃中,并将其缓慢滴加如反应器中;最后在室温搅拌反应30分钟后,薄层色谱监测反应进程,直至反应完全;加入5ml水,用10ml乙酸乙酯分三次萃取,合并乙酸乙酯溶液,用无水硫酸钠干燥,减压蒸出乙酸乙酯,剩余物用石油醚/乙酸乙酯=60/1的流动相经硅胶柱层析分离纯化,得到多取代吡咯化合物iiib,产率82%。
1hnmr(400mhz,cdcl3)δ7.45(dd,j=8.6,5.4hz,2h),7.22(s,5h),7.00(t,j=8.7hz,2h),6.37(d,j=2.0hz,1h),6.30(d,j=1.9hz,1h),3.48–3.34(m,2h),2.87-2.78(t,j=14.7hz,2h),2.19–2.11(m,1h),2.04–1.95(m,1h);13cnmr(101mhz,cdcl3)δ161.7,144.1,135.0,131.3,130.5,130.4,128.9,128.6,127.1,125.8,115.8,115.5,96.6,89.3,77.5,77.1,76.9,28.2,27.9,24.1.
实施例3
在10毫升圆底烧瓶中依次加入化合物ia(0.1mmol)、iic(0.15mmol)在氩气保护下;然后将叔丁醇钾(0.20mmol)溶解在无水的四氢呋喃中,并将其缓慢滴加如反应器中;最后在室温搅拌反应30分钟后,薄层色谱监测反应进程,直至反应完全;加入5ml水,用10ml乙酸乙酯分三次萃取,合并乙酸乙酯溶液,用无水硫酸钠干燥,减压蒸出乙酸乙酯,剩余物用石油醚/乙酸乙酯=60/1的流动相经硅胶柱层析分离纯化,得到多取代吡咯化合物iiic,产率77%。
1hnmr(400mhz,cdcl3)δ7.62-7.56(m,j=8.4hz,4h),7.23(s,5h),6.39(d,j=1.8hz,1h),6.36(d,j=1.8hz,1h),3.48–3.34(m,2h),2.88-2.81(t,j=13.8hz,2h),2.18-2.15(m,1h),2.05-1.99(m,1h);13cnmr(101mhz,cdcl3)δ143.7,142.9,131.0,129.2,128.9,128.6,128.3,127.0,126.0,125.6,96.9,89.2,77.3,77.0,76.7,28.1,27.8,23.9.
实施例4
在10毫升圆底烧瓶中依次加入化合物ib(0.1mmol)、iid(0.15mmol)在氩气保护下;然后将叔丁醇钾(0.20mmol)溶解在无水的四氢呋喃中,并将其缓慢滴加如反应器中;最后在室温搅拌反应30分钟后,薄层色谱监测反应进程,直至反应完全;加入5ml水,用10ml乙酸乙酯分三次萃取,合并乙酸乙酯溶液,用无水硫酸钠干燥,减压蒸出乙酸乙酯,剩余物用石油醚/乙酸乙酯=60/1的流动相经硅胶柱层析分离纯化,得到多取代吡咯化合物iiid,产率75%。
1hnmr(400mhz,cdcl3)δ7.42(dd,j=8.7,5.4hz,2h),7.36(d,j=8.6hz,2h),7.26(s,1h),7.08(d,j=8.6hz,2h),7.01(t,j=8.7hz,2h),6.39(d,j=2.0hz,1h),6.26(d,j=2.0hz,1h),3.48–3.34(m,2h),2.89–2.79(m,2h),2.18–2.13(m,1h),2.05–1.97(m,1h);13cnmr(101mhz,cdcl3)δ161.7,143.0,134.6,131.8,130.5,130.4,130.2,128.6,126.4,123.1,115.9,115.7,96.6,89.1,77.4,77.1,76.8,28.2,27.9,24.0.
实施例5
在10毫升圆底烧瓶中依次加入化合物ic(0.1mmol)、iie(0.15mmol)在氩气保护下;然后将叔丁醇钾(0.20mmol)溶解在无水的四氢呋喃中,并将其缓慢滴加如反应器中;最后在室温搅拌反应30分钟后,薄层色谱监测反应进程,直至反应完全;加入5ml水,用10ml乙酸乙酯分三次萃取,合并乙酸乙酯溶液,用无水硫酸钠干燥,减压蒸出乙酸乙酯,剩余物用石油醚/乙酸乙酯=60/1的流动相经硅胶柱层析分离纯化,得到多取代吡咯化合物iiie,产率73%。
1hnmr(400mhz,cdcl3)δ7.46–7.43(m,2h),7.12(d,j=8.1hz,2h),7.04–6.97(m,4h),6.33(d,j=1.8hz,1h),6.28(d,j=2.1hz,1h),3.49–3.34(m,2h),2.88–2.78(m,2h),2.27(s,3h),2.19–2.11(m,1h),2.04–1.95(m,1h);13cnmr(101mhz,cdcl3)δ161.6,144.0,139.0,135.1,130.5,130.4,129.3,128.4,127.0,124.8,115.7,115.5,96.5,89.3,77.5,77.1,76.8,28.3,27.9,24.1,21.4.
实施例6
在10毫升圆底烧瓶中依次加入化合物id(0.1mmol)、iia(0.15mmol)在氩气保护下;然后将叔丁醇钾(0.20mmol)溶解在无水的四氢呋喃中,并将其缓慢滴加如反应器中;最后在室温搅拌反应30分钟后,薄层色谱监测反应进程,直至反应完全;加入5ml水,用10ml乙酸乙酯分三次萃取,合并乙酸乙酯溶液,用无水硫酸钠干燥,减压蒸出乙酸乙酯,剩余物用石油醚/乙酸乙酯=60/1的流动相经硅胶柱层析分离纯化,得到多取代吡咯化合物iiif,产率75%。
1hnmr(400mhz,cdcl3)δ7.47(d,j=6.8hz,1h),7.37–7.29(m,4h),7.17(d,j=6.8hz,2h),6.72(d,j=8.8hz,2h),6.26(dd,j=5.3,1.9hz,2h),3.71(s,3h),3.41(m,2h),2.81(m,2h),2.13(m,1h),1.98(m,1h);13cnmr(101mhz,cdcl3)δ159.9,143.7,139.3,128.8,128.7,128.6,123.9,123.6,113.9,96.6,90.1,77.5,77.2,76.8,55.3,28.3,28.0,24.2
- 还没有人留言评论。精彩留言会获得点赞!