一种用于免疫分析的高效氯氟氰菊酯半抗原、活化载体蛋白、完全抗原及多克隆抗体的制作方法
本发明涉及检测技术领域,尤其涉及一种用于免疫分析的高效氯氟氰菊酯半抗原、活化载体蛋白、完全抗原及多克隆抗体。
背景技术:
拟除虫菊酯类杀虫剂是一类模拟天然除虫菊酯的合成杀虫剂,是一种由菊酯生物合成的昆虫神经毒素。到目前为止,拟除虫菊酯已经占据了全球农药市场18%以上的份额。高效氯氟氰菊酯(如下文式1所示)是该类杀虫剂的重要组成之一,在农业生产中有着广泛的应用。尽管人们普遍认为它的毒性很低,但最近的两项研究结果对这种分子对人体的安全性提出了重大质疑。例如,研究表明,过度暴露于氰菊酯可能导致肌痛性、急性呼吸道刺激性损伤和休克。另一方面,由于在蔬菜、水果和鱼类等食品中发现高效氯氟氰菊酯残留,使人接触到氰菊酯的风险大大增加。为了控制人类对该杀虫剂的接触,许多国家和地区都对高效氯氟氰菊酯残留有严格的规定。例如,中国和欧盟食品中高效氯氟氰菊酯的最大残留限量分别为0.05mg/kg~15mg/kg和0.02mg/kg~1mg/kg。
目前,食品中菊酯类化合物(包括高效氯氟氰菊酯)的残留分析主要采用高效液相色谱法(hplc)或gc-ms法。尽管这些仪器分析方法具有较高的准确度和较低的检出限(lod),但由于成本高(尤其是在较贫穷的国家)、样品制备复杂、与现场检测不兼容(如农市场)等原因,这些方法的广泛应用受到了影响。与仪器方法相比,免疫分析法更便宜、更简便,尤其适合现场检测,因此受到了学术界和工业界的广泛关注。免疫检测方法的发展一般遵循半抗原合成、半抗原蛋白偶联和抗体形成三个步骤,其中半抗原合成通常被认为是关键步骤。在高效氯氟氰菊酯的半抗原合成中,虽然已经发表了多种合成方法,但是它们的合成路线仍然有优化的空间(如下文所示)。例如,gao等报道了拟除虫菊酯半抗原的多步合成,但该方法存在高毒性试剂(其他一些拟除虫菊酯半抗原合成方法也存在此问题,例如c中lee等报道的方法),总收率低(10.4%)的问题。luo和其同事报道了使用无毒kmno4氧化一步合成高效氯氟氰菊酯半抗原的方法,但是这种方法去掉了区分不同拟除虫菊酯的关键官能基(-cf3和-cl),得到的半抗原的结构过于普遍,因此被认为是非特异性的。
技术实现要素:
为了解决上述技术问题,本发明利用低毒性试剂选择性水解氰基,可一步合成高效氯氟氰菊酯半抗原。将该带酰胺基的半抗原与琥珀酸酐活性载体蛋白结合形成完全抗原,后者用于生成多克隆抗体。该抗体对高效氯氟氰菊酯的最低检出限为10μg/l,对其他拟除虫菊酯类农药(溴氰菊酯、氯氰菊酯、氟氯氰菊酯和氰戊酸酯)无明显交叉反应。
本发明的具体技术方案为:
第一方面,本发明提供了一种用于免疫分析的高效氯氟氰菊酯半抗原,其分子结构式为:
第一方面,本发明提供了一种高效氯氟氰菊酯半抗原的一步合成方法,其化学反应过程如下所示:
在半抗原的合成方面:本发明为了构建具有高特异性的高效氯氟氰菊酯抗原,第一个要求是设计一个简单和安全的半抗原。本发明直接从高效氯氟氰菊酯进行修改(它包含所有的结构信息,而且相当便宜)以达到最高效。并且坚持只使用非有毒试剂或低毒试剂。此外,我们更倾向于对杀虫剂结构进行最小的修改,以确保最终抗原的识别和特异性。
氰基在效氯氟氰菊酯的中心部分是一个多功能基团,可以转化成许多其他基团。因此,发明人在研发过程中原先是考虑利用三甲基氯硅烷(tmscl)原位生成的hcl(pka=-5.9)和h2o催化水解效氯氟氰菊酯。然而,最初使用tmscl作为催化剂的尝试未能生成产物。然后,我们假设反应失败是由于酸度不足造成的,并切换了三甲基硅基三氟甲烷磺酸盐(tmsotf),它与水反应时产生更酸性的tfoh(pka=-14.7)。结果非常理想,反应产生了产率为69%的理想半抗原。与背景技术部分中介绍的gao等人的5步合成高效氯氟氰菊酯半抗原相比,本发明方法更高效和更容易工业化生产。此外,该改造过程保留了大部分官能团的完整性(如-cf3和-f),它们比背景技术部分中介绍的luo等人合成的半抗原具有更高的结构保真度,因此更具特异性。
作为优选,原料以mmol计,一步合成方法具体包括以下步骤:将1.5-2.5mmol高效氯氟氰菊酯装入反应容器中在冰水浴中冷却,然后加入3.5-4.5mmol三甲基((三氟甲基)磺酰)硅烷,搅拌均匀;待高效氯氟氰菊酯完全溶解后,缓慢加水并对所得混合物进行搅拌;然后添加试剂对反应进行中和,用有机试剂萃取;将有机相混合并使用脱水试剂进行干燥,然后真空浓缩;产物使用硅胶柱分离,淋洗液为乙酸乙酯和正己烷的混合液。
作为优选,加入1三甲基((三氟甲基)磺酰)硅烷后搅拌1-3min。
作为优选,将70-80μl的水在1.5-2.5min内缓慢加入,对所得混合物搅拌3-5h。
作为优选,用饱和的nahco3溶液对反应进行中和。
作为优选,用有机试剂ch2cl2萃取多次,每次用量为10-20ml。
作为优选,将有机相混合并使用过量的na2so4干燥。
作为优选,所述淋洗液中乙酸乙酯和正己烷的体积比为(35-65)∶(45-55)。
第三方面,本发明提供了一种活化载体蛋白的制备方法,包括以下步骤:
将载体蛋白ova在双蒸水中溶解,调节ph至8.0~8.5;然后缓慢加入琥珀酸酐;将ph=8.0~9.0的混合溶液进行搅拌;用磷酸缓冲液透析,然后将产物保存。
第四方面,本发明提供了一种用于免疫分析的高效氯氟氰菊酯完全抗原的制备方法,包括以下步骤:
1)载体蛋白活化:将载体蛋白ova在双蒸水中溶解,调节ph至8.0~8.5;然后缓慢加入琥珀酸酐;将ph=8.0~9.0的混合溶液进行搅拌;用磷酸缓冲液透析,然后将产物保存。
2)半抗原蛋白偶联:将权利要求1所述的高效氯氟氰菊酯半抗原或权利要求2-4之一所述方法所得的高效氯氟氰菊酯半抗原通过碳二亚胺法与活化载体蛋白偶联;得到半抗原-ova偶联物,即高效氯氟氰菊酯完全抗原。
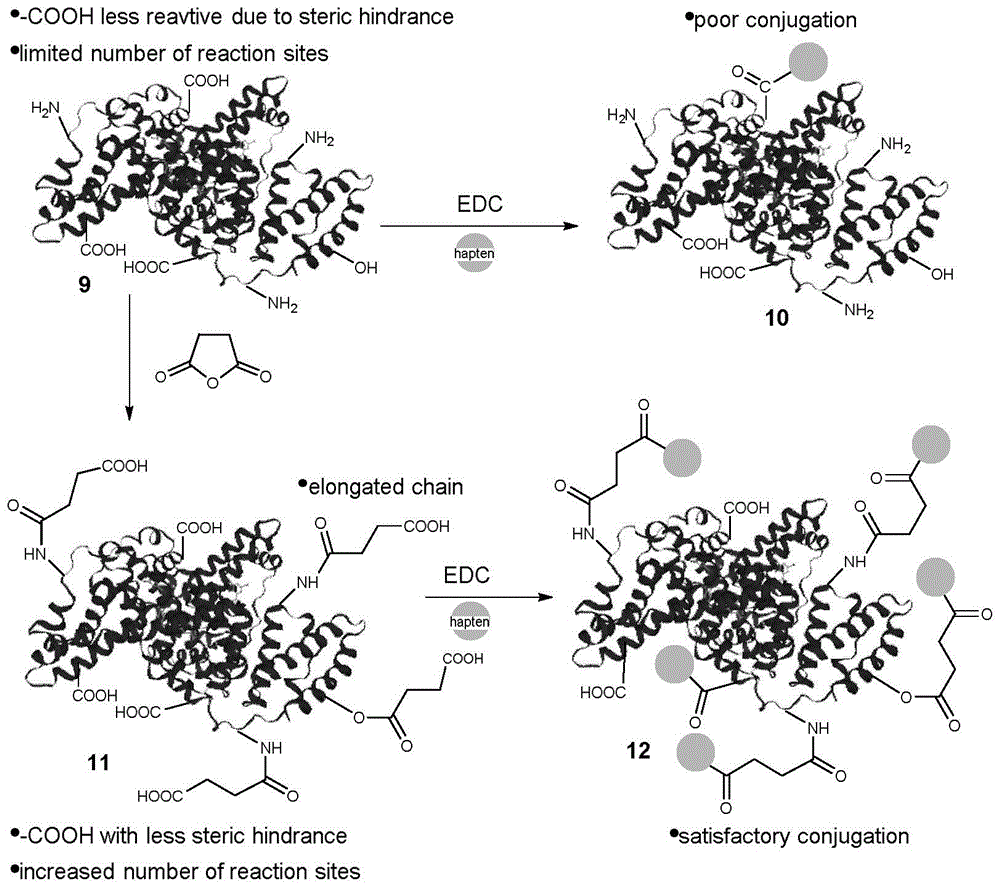
在载体蛋白活化与半抗原结合方面:为了产生抗体,半抗原必须与载体蛋白结合。因此,高效氯氟氰菊酯半抗原使用edc方法与bsa和ova上的羧酸基团结合。然而,紫外光谱定性分析显示偶联效果不佳。本发明团队经过研究,发现这是因为酰胺基对羧酸的反应活性要比氨基小得多,而且载体蛋白上的天然氨基可以与羧酸发生竞争性反应。此外,载体蛋白中的羧酸可能被近端结构部分甚至完全阻断,从而影响其反应活性。并且,由于bsa和ova是不特别富含asp和glu残基的蛋白,羧基的含量不高。为了提高偶联率,本发明最终用琥珀酸酐激活载体蛋白。该反应将蛋白质表面的氨基和羟基分别转化为琥珀酸单酰胺和酯基,同时生成羧基。因此,该处理将额外的羧基基团引入到载体蛋白中,这些羧基基团预期比天然的羧基基团反应性更强,因为它们被设计成从蛋白体中释放出来,因此阻碍更小。此外,该反应将氨基基团聚集在载体蛋白上,抑制竞争性的酰胺化反应。
作为优选,所述载体蛋白在双蒸水中的浓度为45-55mg/ml。
作为优选,所述琥珀酸酐的加入量为载体蛋白质量的10-20%。
作为优选,所述混合溶液在20-30℃下连续搅拌2-4h。
作为优选,所述磷酸缓冲液中含0.05-0.15mol/l的nacl。
第五方面,本发明提供了一种用于免疫分析的高效氯氟氰菊酯多克隆抗体的制备方法,包括以下步骤:对一批小鼠腹腔分多次注射高效氯氟氰菊酯完全抗原;其中,第一次注射的同时注射完全弗氏佐剂;后续的注射用不完全弗氏佐剂代替完全弗氏佐剂;除首次注射外,小鼠均于每次注射后采血,以监测抗血清的效价;用酶联免疫吸附法测定各小鼠抗血清的效价,根据性能指标选择效价最高的抗血清,用蛋白a柱进一步纯化,最后将所得多克隆抗体储存备用。
本发明通过免疫小鼠产生抗体。经过多轮免疫后,16种抗血清的效价均超过20万,其中12只在1mg/kg的高效氯氟氰菊酯存在下抑制率超过30%,说明抗血清的产生并非偶然。这些结果也表明抗血清与高效氯氟氰菊酯具有显著的特异性结合能力,从而证实了半抗原设计和抗体产生的成功。
此外,本发明对抗体进行的纯化处理。通过ic-elisa绘制标准的抑制曲线。ic50值大约为50μg/l和最低检测极限(lod)(ic10)大约10μg/l。在世界上许多地区,食物中高效氯氟氰菊酯的最大残留限量范围在50μg/kg~15mg/kg之间,这高于免疫测定的lod值。因此,如果对不同的样品进行有效的预处理,制备的抗体对高效氯氟氰菊酯的灵敏度是令人满意的。
作为优选,所述一批小鼠为10-20只6~8周的雌性balb/c小鼠。
作为优选,所述分多次注射为分4-6次注射高效氯氟氰菊酯完全抗原,每次间隔8-12天;每次注射150-250μl浓度为0.8-1.2毫克/毫升的高效氯氟氰菊酯完全抗原以及150-250μl完全弗氏佐剂或不完全弗氏佐剂。
作为优选,所述采血为每次注射后5-9天从小鼠尾静脉采血。
与现有技术对比,本发明的有益效果是:
(1)本发明利用低毒性试剂选择性水解氰基,可一步合成高效氯氟氰菊酯半抗原。与现有技术中的多步合成高效氯氟氰菊酯半抗原相比,本发明方法更高效和更容易工业化生产。此外,该改造过程保留了大部分官能团的完整性(如-cf3和-f),它们比现有技术中的半抗原具有更高的结构保真度,因此更具特异性。
(2)本发明在将高效氯氟氰菊酯半抗原与载体蛋白结合前,先将载体蛋白被琥珀酸酐活化,琥珀酸酐不仅提供了较少受阻的额外羧基端,而且减弱了天然氨基的竞争性偶联。
(3)本发明再将高效氯氟氰菊酯半抗原与琥珀酸酐活性载体蛋白结合形成完全抗原,后者用于生成多克隆抗体。该抗体对高效氯氟氰菊酯的ic50和lod分别评价为50μg/l和10μg/l,对其他拟除虫菊酯类农药(溴氰菊酯、氯氰菊酯、氟氯氰菊酯和氰戊酸酯)无明显交叉反应,显示出极强的特异性。
附图说明
图1为载体蛋白与半抗原的不同方式结合图示;
图2为抗血清对1mg/kg的氟氯氰菊酯的抑制率图;
图3为优化后的高效氯氟氰菊酯酶联免疫吸附试验标准曲线;其中:包被抗原浓度5μg/l;抗体稀释1/100000;羊抗鼠二抗稀释,1/5000;ph值为7.0。
具体实施方式
下面结合实施例对本发明作进一步的描述。
总实施例
用于免疫分析的高效氯氟氰菊酯半抗原,其分子结构式为:
高效氯氟氰菊酯半抗原的一步合成方法,其化学反应过程如下所示:
原料以mmol计,一步合成方法具体包括以下步骤:将1.5-2.5mmol高效氯氟氰菊酯装入反应容器中在冰水浴中冷却,然后加入3.5-4.5mmol三甲基((三氟甲基)磺酰)硅烷,搅拌1-3min至均匀;待高效氯氟氰菊酯完全溶解后,在1.5-2.5min内缓慢加70-80μl水并对所得混合物进行搅拌3-5h;然后用饱和的nahco3溶液对反应进行中和,用有机试剂ch2cl2萃取多次,每次用量为10-20ml;将有机相混合并使用使用过量的na2so4进行干燥,然后真空浓缩;产物使用硅胶柱分离,淋洗液为体积比为(35-65)∶(45-55)的乙酸乙酯和正己烷的混合液。
高效氯氟氰菊酯完全抗原的制备方法,包括以下步骤:
1)载体蛋白活化:将载体蛋白ova以浓度45-55mg/ml在双蒸水中溶解,调节ph至8.0~8.5;然后缓慢加入琥珀酸酐至其质量为载体蛋白的10-20%;将ph=8.0~9.0的混合溶液在20-30℃下连续搅拌2-4h;用磷酸缓冲液透析,然后将产物保存。
2)半抗原蛋白偶联:将高效氯氟氰菊酯半抗原通过碳二亚胺法与活化载体蛋白偶联;得到半抗原-ova偶联物,即高效氯氟氰菊酯完全抗原。
高效氯氟氰菊酯多克隆抗体的制备方法,包括以下步骤:
对10-20只6~8周的雌性balb/c小鼠腹腔分4-6次注射高效氯氟氰菊酯完全抗原,每次间隔8-12天;150-250μl浓度为0.8-1.2毫克/毫升的高效氯氟氰菊酯完全抗原以及150-250μl完全弗氏佐剂或不完全弗氏佐剂。其中,第一次注射的同时注射完全弗氏佐剂;后续的注射用不完全弗氏佐剂代替完全弗氏佐剂;除首次注射外,小鼠均于每次注射后5-9天从小鼠尾静脉采血,以监测抗血清的效价;用酶联免疫吸附法测定各小鼠抗血清的效价,根据性能指标选择效价最高的抗血清,用蛋白a柱进一步纯化,最后将所得多克隆抗体储存备用。
具体实施例
材料
试剂:二氧化硅凝胶(125~150μm)是购自上海阿拉丁生化科技有限公司有限公司(中国上海);高效氯氟氰菊酯购自武汉元成共超科技有限公司(中国武汉);溴氰菊酯、氯氰菊酯、氟氯氰菊酯和氰戊菊酯均来自中国国家标准物质研究中心(北京,中国)。脱脂奶粉购自reanal(匈牙利)。半抗原合成及与载体蛋白偶联的化学物质,辣根过氧化物酶(hrp)、牛血清白蛋白(bsa)、卵清蛋白(ova)和四甲基联苯胺(tmb)偶联的山羊抗小鼠igg购自sigma-aldrichchemicalco.(密尔沃基,美国)。α-cyano-4hydroxycinnamic-acid(hcca),三氟乙酸(tfa)购自sigma-aldrich化工有限公司(密尔沃基,美国)。
仪器:酶联免疫吸附试验(elisas)在96孔板(3655号)中进行,这些微效板购自thermofisher科学公司(马萨诸塞州沃尔瑟姆,美国),用酶标仪读取吸光度。质谱和核磁共振(nmr)分析在青岛海洋生物医学研究所进行:uhplc-三重四极质谱仪(1290infinityiiuhplc/6460qqqms)和核磁共振光谱仪(propulse500mhz)均来自安捷伦(美国)。核磁共振波谱仪(propulse500mhz,agilent,usa)。maldi-tof分析在齐鲁理工大学(山东科学院)山东分析测试中心进行:rapiflexmaldi-tof/tof来自德国bruker公司。所有数据由originlab(马萨诸塞州北安普顿,美国)的origin9.0、cambridgesoft(马萨诸塞州,美国)的chemdraw和chem3d制作,使用swiss-model模拟蛋白结构。
1hnmr:化学变化调整如下:δh(chcl3)=7.27ppm。在本发明中,1hnmr化学位移以ppm表示,小数点后两位;多重性被描述为:“s”表示siglet,“d”表示doublet,“t”表示trplet,“q”表示quartet,“p”表示pentet,“m”表示multiplet,“br”表示broad。
方法
(1)半抗原的合成:一个装有0.900g(2mmol,1equiv)的25ml烘干圆底烧瓶在0℃冰水浴中冷却。然后加入825mg三甲基((三氟甲基)磺酰)硅烷(me3siso2cf3,4mmol,2equiv.),搅拌2分钟。高效氯氟氰菊酯完全溶解后,72μl的水在2分钟内慢慢加入,混合物搅拌4h。然后用饱和的nahco3溶液对反应进行中和,用ch2cl2萃取两次(15mlx2)。将有机相混合并使用过量的na2so4干燥,然后在真空中浓缩。产物使用硅胶柱分离(125~150μm)淋洗液为乙酸乙酯和正己烷(40∶60)。
所得产物为一种无色液体(643mg,产率为69%)。
1hnmr(500mhz,cdcl3,298k):h7.37-7.34(3h,m,ar-h),7.22(1h,d,j=7.5hz,ar-h),7.15-7.12(2h,m,ar-h),7.03-6.98(3h,m,ar-h),6.16(1h,d,j=8.5hz,=ch),6.06(1h,s,ochcn),6.02(1h,br,-nh),5.56(1h,br,-nh),2.43(1h,dd,j=9.0hz,5.0hz,=ch-ch),1.89(1h,d,j=5.0hz,ch-co2),1.36(3h,s,ch3),1.29(3h,s,ch3).ms(esi+):490.3[m+na]+.
(2)载体蛋白活化:载体蛋白(bsa或ova)以50mg/ml的浓度在双蒸水中溶解,naoh(0.2mol/l)调节ph至8.0~8.5。然后缓慢加入琥珀酸酐,使其达到载体蛋白质量的15%。混合溶液(ph=8.0~9.0)在25℃下连续搅拌3小时。反应后用考马斯亮蓝法测定蛋白浓度。用磷酸缓冲液(0.1mol/l,含0.1mol/lnacl,pbs)透析72h,-20℃保存。
(3)半抗原蛋白偶联:半抗原通过碳二亚胺法与活化载体蛋白偶联。用maldi-tof/ms对制备的偶联物进行分析。基质辅助溶液hcca在ta中饱和(acn:0.1%tfa,1∶2)。将脱盐后的样品溶解在0.1%tfa中。1μl样本加到靶标(384不锈钢)上并在室温下干燥。然后添加1μlhcca基质并干燥。检测条件为:激光照射:5000次;采样率:1.25gss-1;加速电位:20kv,真空压力:3~510-7mbar;脉冲离子提取延迟:100纳秒。结合物的偶联率计算如下:
其中,所得半抗原-ova偶联物作为免疫原,半抗原-bsa偶联物作为包被抗原(用于步骤(5))。
(4)多克隆抗体生产:16只6~8周的雌性balb/c小鼠分五次腹腔注射200μl半抗原-ova偶联物(1毫克/毫升),每次间隔10天。另取4只6~8周的雌性balb/c小鼠,每隔10天腹腔注射相同剂量的生理盐水5次,作为阴性对照。其中,第一次注射半抗原的同时注射200μl完全弗氏佐剂。后续的注射用不完全佐剂代替完全佐剂。除首次注射外,小鼠均于每次注射后7天从尾静脉采血,以监测抗血清的效价。用酶联免疫吸附法测定各小鼠抗血清的效价。根据性能指标选择效价最高的抗血清,用蛋白a柱进一步纯化。然后将制备好的抗体储存在-20℃,以备进一步使用。
(5)elisa程序:采用双抗体夹心elisa法检测其效价,方法与ferencszurdoki等报道的方法相同。96孔微量滴定板中加入10μg/毫升溶解在碳酸缓冲溶液(0.1m,ph值9.6,cbs)中的包被抗原,在37℃下孵育2小时,然后用5%脱脂牛奶的pbst溶液4摄氏度下过夜孵育。用甲醇-pbs混合液(50∶50,v∶v)将抗血清稀释至不同浓度,加入孔中,37℃孵育1.5小时。pbst洗3次,每个微孔加入100μl羊抗鼠免疫球蛋白辣根过氧化物酶共轭物(pbs1∶5000稀释),37℃孵育2小时。pbst洗3次,每个微孔加入100μl底物显色液,37℃孵育5~10分钟,在492nm下测定光密度值(od)。elisa中阴性对照用阴性血清代替抗血清,空白对照用pbs代替抗血清。
间接竞争elisa(ic-elisa)中,以甲醇溶解的5mg/l的高效氯氟氰菊酯为原液,用50%甲醇-pbs连续稀释。将梯度稀释至不同浓度的原液与等量的抗体混合。37℃中孵育5分钟后,将混合物加入微孔中。其他步骤与双抗体夹心elisa相同。利用常用的四参数曲线拟合方法生成s形标准曲线。四参数logistic方程如下
ic50表示50%抑制所需的最低分析物浓度。低检测限(ldl)被定义为10%抑制所需的最低分析物浓度。ic50值和ldl均通过logistic方程得到。
用ic-elisa法测定制备的抗体对其他农药(溴氰菊酯、氯氰菊酯、氟氯菊酯和氰戊酸酯)的特异性。用化合物最低浓度达到50%抑制(ic50)计算交叉反应活性(cr),公式如下
结果与讨论
半抗原的合成:本发明的一个主要目标是构建具有高特异性的高效氯氟氰菊酯抗原,该抗原具有工业化的潜力。为了实现这一目标,第一个要求是设计一个简单和安全的半抗原的合成,我们决定直接从高效氯氟氰菊酯进行修改(它包含所有的结构信息,而且相当便宜)以达到最高效。并且坚持只使用非有毒试剂或低毒试剂。此外,我们更倾向于对杀虫剂结构进行最小的修改,以确保最终抗原的识别和特异性。
氰基在效氯氟氰菊酯的中心部分是一个多功能基团,可以转化成许多其他基团。因此,我们在研发过程中原先是考虑利用三甲基氯硅烷(tmscl)原位生成的hcl(pka=-5.9)和h2o催化水解效氯氟氰菊酯。然而,最初使用tmscl作为催化剂的尝试未能生成产物。然后,我们假设反应失败是由于酸度不足造成的,并切换了三甲基硅基三氟甲烷磺酸盐(tmsotf),它与水反应时产生更酸性的tfoh(pka=-14.7)。结果非常理想,反应产生了产率为69%的理想半抗原。与背景技术部分中介绍的gao等人的5步合成高效氯氟氰菊酯半抗原相比,本发明方法更高效和更容易工业化生产。此外,该改造过程保留了大部分官能团的完整性(如-cf3和-f),它们比背景技术部分中介绍的luo等人合成的半抗原具有更高的结构保真度,因此更具特异性。
载体蛋白活化与半抗原结合:为了产生抗体,半抗原必须与载体蛋白结合。因此,高效氯氟氰菊酯半抗原使用edc方法与bsa和ova上的羧酸基团结合(图1中的9→10)。然而,紫外光谱定性分析显示偶联效果不佳。这个不受欢迎的结果并不奇怪,因为酰胺基对羧酸的反应活性要比氨基小得多,而且载体蛋白上的天然氨基可以与羧酸发生竞争性反应。此外,载体蛋白中的羧酸可能被近端结构部分甚至完全阻断,从而影响其反应活性。此外,由于bsa和ova是不特别富含asp和glu残基的蛋白,羧基的含量不高。为了提高偶联率,我们用琥珀酸酐激活载体蛋白(图1中的9→11)。该反应将蛋白质表面的氨基和羟基分别转化为琥珀酸单酰胺和酯基,同时生成羧基。因此,该处理将额外的羧基基团引入到载体蛋白中,这些羧基基团预期比天然的羧基基团反应性更强,因为它们被设计成从蛋白体中释放出来,因此阻碍更小。此外,该反应将氨基基团聚集在载体蛋白上,抑制竞争性的酰胺化反应。
如图1所示,活化后的载体蛋白11与半抗原反应,得到满意的偶联。为了计算平均耦合产生的完全抗原,随样品进行maldi-tof检测,结果表明半抗原对bsa和ova的偶联比分别为15∶1和10∶1。半抗原与载体蛋白的低偶联在合成完全抗原的步骤中是一个常见问题的。解决这一问题的方法主要集中在半抗原的构建上,引入氨基或羧基是主要的趋势。本发明为载体蛋白引入反应性官能团,为完全抗原的合成带来更大的灵活性提供了一个范例。更重要的是,由此产生的修饰蛋白有可能被标准化,甚至作为更活跃的载体进行商业化,而且这种反应至少在理论上是可控的。
抗体的产生和表征:免疫16只雌性balb/c小鼠产生抗体。经过5轮免疫后,16种抗血清的效价均超过20万,其中12只在1mg/kg的高效氯氟氰菊酯存在下抑制率超过30%(图2),说明抗血清的产生并非偶然。这些结果也表明抗血清与高效氯氟氰菊酯具有显著的特异性结合能力,从而证实了半抗原设计和抗体产生的成功。
纯化抗体与抗血清:通过ic-elisa绘制标准的抑制曲线(图3)。ic50值大约为50μg/l和最低检测极限(lod)(ic10)大约10μg/l。在世界上许多地区,食物中高效氯氟氰菊酯的最大残留限量范围在50μg/kg~15mg/kg之间,这高于免疫测定的lod值。因此,如果对不同的样品进行有效的预处理,制备的抗体对高效氯氟氰菊酯的灵敏度是令人满意的。
通过与其他拟除虫菊酯的交叉反应来评价制备的抗体的特异性。如表1所示,与高效氯氟氰菊酯(100%)相比,其他待测物的交叉反应活性均在10%以下,这表明生成的抗体具有良好的选择性。特别是,氯氰菊酯只是一个不同于高效氯氟氰菊酯的官能团(clvscf3),而半抗原仅展现出了5%的交叉反应活性。这一结果表明,高效氯氟氰菊酯中的所有官能团都参与了抗体-抗原的识别过程。
表1不同拟除虫菊酯间交叉反应率测定。
结论
本发明利用tmsotf催化氰胺水解反应合成了一种新型的高效氯氟氰菊酯半抗原。载体蛋白被琥珀酸酐活化,琥珀酸酐不仅提供了较少受阻的额外羧基端,而且减弱了天然氨基的竞争性偶联。用活化的载体蛋白和新的半抗原产生完全抗原,并从小鼠体内产生多克隆抗体,对高效氯氟氰菊酯的ic50和lod分别评价为50μg/l和10μg/l,对其他拟除虫菊酯无明显交叉反应。因此我们认为这一研究表明,一步合成策略比现有的方法更容易,更安全,产量更高,这可能是合成带氰基农药半抗原的一个方便的方法。此外,载体蛋白活化方法为低结合问题提供了一种新的解决方案,有望成为一系列标准化活化载体蛋白的原型。
本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!