一种葡萄糖敏感的苯硼酸衍生物的合成及表征
本发明涉及一种新结构类型的苯硼酸衍生物的合成及表征,尤其是涉及apcpba、apcfpba及apccf3pba的化学合成、制备方法及其结构表征,并且可以应用于载胰岛素的释放体系中,属于化学合成
技术领域:
。
背景技术:
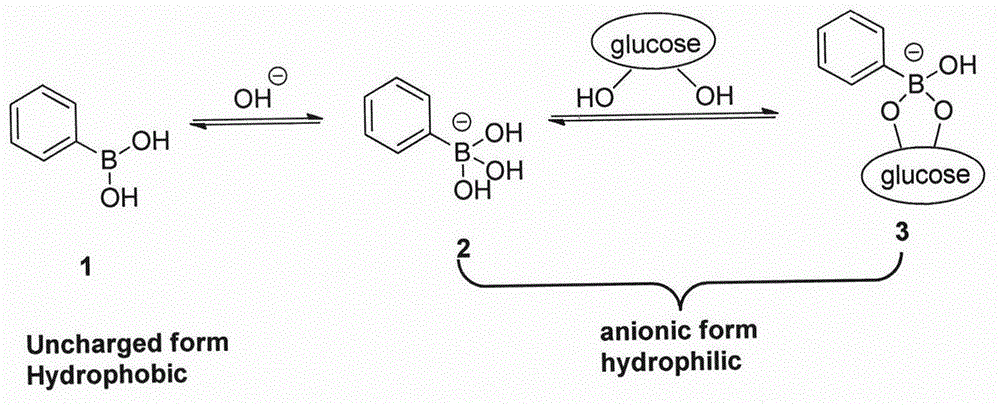
:糖尿病是由于胰腺分泌的胰岛素不足而引起的一种新陈代谢疾病,严重威胁人类健康。目前医学上还无法根治,只能通过控制病人的血糖水平来推迟并发症的出现。因此对于糖尿病的控制和治疗来说,研制能够感知体内血糖浓度变化的智能材料是十分有意义的。这样的材料能够实时评估检测病人的血糖浓度,及时反馈病情甚至根据血糖水平自动调整释放药物。不间断的测定血糖浓度并依糖释放胰岛素是控制糖尿病的有效方法。目前被研究的糖敏药物缓控释材料主要有三类:第一类是将葡萄糖氧化酶(god)包埋在ph敏感水凝胶中构成的糖敏水凝胶体系。在氧气存在的条件下,god会降解周围环境中的葡萄糖催化氧化为葡萄糖酸,从而降低体系的ph值,导致水凝胶溶胀度的增大或减小;第二类是以一种外源凝集素--伴刀豆球蛋白(cona)为糖敏感基元的材料。cona分子上有可与糖基结合的位点,将cona与糖基化药物的复合物置于葡萄糖环境下,葡萄糖会与糖基化药物竞相与cona反应,替换下药物。以上两类糖敏智能体系虽具有较好的葡萄糖敏感性,但它们是基于蛋白质的体系,储存时间短,稳定性差,并具有一定的毒性。相比之下,使用苯硼酸(pba)为糖敏感基元的第三类材料是合成体系,稳定性好,不会产生免疫排斥作用。pba及其衍生物是疏水单体,与葡萄糖形成复合物后呈现出亲水性,这种转变可以引起聚合物体系溶胀度的改变。这类物质在水溶液中以两种形式存在,一种是亲水性的带负电荷的解离态,另一种是疏水性的不带电荷的未解离态,两种状态之间存在解离平衡,这两种形态下解离的pba更容易通过共价键结合葡萄糖,形成更加亲水的结构,并促进电离平衡向右移动(图1)。将pba或其衍生物制备成聚合物凝胶,在非葡萄糖环境中,苯硼酸基团的疏水作用占主导,凝胶处于收缩状态,随着环境中葡萄糖浓度的升高,解离的苯硼酸基团与葡萄糖反应,亲水性增强,使凝胶具有良好的水化作用而逐渐溶胀(图2),因此pba的这种葡萄糖响应性可以用来设计自调节胰岛素释放体系,用于血糖的检测和控制。尽管优势明显,但由于苯硼酸是一个lewis酸,在构筑葡萄糖响应性聚合物材料时存在一个缺陷就是它的pka(7.8-8.6)比生理ph(7.2-7.4)高很多,在生理条件下,大部分苯硼酸都处于未电离状态,而未电离状态和葡萄糖结合作用力很弱,响应能力很低。研究人员为了提高苯硼酸在生理条件下的响应能力尝试了很多方法,一是在苯硼酸上引入吸电子基团,如在苯硼酸对位上引入羧基或是间位上引入卤素或硝基,二是在硼酸邻位引入氨基或是羟基等配位基团,和硼原子配位以调节到合适的pka。因此本领域还需要有新的结构类型的苯硼酸衍生物作为葡萄糖敏感的基元分子,在生理条件下作为载药释放的载体,为人们提供一种新的安全有效的应用选择。技术实现要素:本发明公开了一种新的结构类型的苯硼酸衍生物的合成及表征。根据文献调研发现的3个具有葡萄糖敏感性的苯硼酸衍生物(图3),本发明在此结构基础上置换连接臂得到新结构类型的苯硼酸衍生物,合成过程经过优化后合成的产率提高,理化性质分析其具有更好的葡萄糖敏感性。本发明的苯硼酸衍生物可以用于胰岛素载药体系的构建,用于治疗糖尿病。本发明的目的在于提供了一种新苯硼酸衍生物的结构及其合成方法,其结构如下所示:其中r取代基可以为h,f,cl,br,i,cf3,ccl3,cbr3,ci3,cn,cooh。本发明的另一目的在于提供了新苯硼酸衍生物的合成方法,其合成过程如下所示:其中r取代基可以为h,f,cl,br,i,cf3,ccl3,cbr3,ci3,cn,cooh。本发明的再一个目的是通过使用这种苯硼酸基元分子的使用,构建的载胰岛素释放体系可以用于治疗糖尿病。本发明的目的是通过如下的技术方案实现的:本发明提供了此类苯硼酸衍生物的合成方法,本发明采用缩合剂完成此类衍生物的酰胺键连接。采用无水boc-哌嗪为起始原料,与丙烯酰氯缩合形成boc-哌嗪丙烯酰胺,而后脱除boc保护基,与取代的苯硼酸缩合得到目标分子。经制备液相纯化冻干得到苯硼酸衍生物的纯品。本发明采用pion/pulse的理化分析仪,对合成得到的苯硼酸衍生物进行pka测试,测定其在生理条件下的葡萄糖的敏感性。以下是本发明中涉及的苯硼酸衍生物的pka测试结果见表1:表1葡萄糖敏感的苯硼酸衍生物的pka值编号pkaddopba7.8amecpba7.2apcpba7.88apcfpba6.82apccf3pba6.71上述测试结果表明在生理条件下apcfpba,apccf3pba将具有较好的葡萄糖敏感性,可作为构建胰岛素载药体系的载体用于治疗糖尿病。以下通过实施例对本发明做进一步描述。附图说明图1pba与葡萄糖反应示意图图2含pba的聚合物凝胶随葡萄糖浓度的增加释放药物图3具有葡萄糖敏感的pba类似物图4说明书摘要附图具体实施方式实施例11.apcpba的具体合成步骤如下所示:(1)boc-哌嗪丙烯酰胺的合成将化合物1,boc-哌嗪(57.0g,306mmol)和et3n(34.0g,336mmol,46.8ml)溶解在500mlch2cl2中,0℃条件下,将化合物1a丙烯酰氯(30.4g,336mmol,27.4ml)滴加到上述溶液中,混合液保持在15℃反应12h。液质监测反应完全后,反应液用水洗(300ml水,分别洗涤2次),水层用二氯甲烷萃取三次(50ml*3),保留有机层,合并后的有机层用无水硫酸钠干燥,旋去溶剂得到棕色油2(80.0g,粗品,1hnmr(cdcl3,400mhz)δ6.52(q,j=6.4hz,1h),6.26(dd,j1=1.6hz,j2=16.8hz,1h),5.70~5.62(m,1h),3.61~3.32(m,8h),1.43(s,9h))。(2)哌嗪丙烯酰胺的合成将化合物2(80.0g,332mmol)溶解在4mhcl的乙酸乙酯溶液中,15℃搅拌5h。液质显示反应完全,反应液过滤后,收集固体。再旋干得到白色固体化合物3(41.0g,292mmol,粗品),直接用于下一步的反应,不进行纯化。(1hnmr(dmso-d6,400mhz)δ7.00(br.s,2h),6.79(m,1h),6.14(dd,j1=2.4hz,j2=16.8hz,1h),5.73(dd,j1=2.4hz,j2=10.4hz,1h),3.80~3.68(m,8h))。(3)apcpba的合成将化合物3(41.0g,232mmol)和化合物3a(38.5g,232mmol)溶解在400mldmf中,并且加入edci(66.7g,348mmol),hobt(47.0g,348mmol),et3n(70.4g,696mmol,96.9ml)。反应液在15℃反应12h。液质结果显示反应完全,反应液用水(600ml)稀释,用hcl(4m,400ml)调节ph至ph=2-3之间。反应液用etoac(400mlx5)萃取,合并有机层,并用旋转蒸发仪旋去溶剂,水相旋干溶剂得到粗产物,再用ch3cn(300ml)悬浮搅拌溶解粗产物30min。混合液过滤,滤饼旋干溶剂得到粗产品,用柱层析色谱(ch2cl2∶ch3oh=10∶1)和制备液相(column:phenomenexlunac18250*50mm*10μm;mobilephase:[water(0.1%tfa)-acn];b%:0%-25%,16min)分离纯化的到目标产物白色固体apcpba(10.5g,36.6mmol,42.2%yield)。(4)apcpba的pka测定称取适量的apcpba或其类似物溶解于40ml的0.01n的naoh(150mmnacl)溶液中,用0.01n的hcl溶液在pka/logp测定仪进行检测,得到pka,已得到的apcpba化合物的pka值为7.88,与母体化合物ddopba的pka值大致相当。(5)结构信息合成的单体化合物经结构表征,包括核磁氢谱、碳谱、质谱、红外、dsc熔点检测,结果如下所示:1hnmr(dmso-d6,400mhz)δ(ppm)8.04(brs,1h),7.84(d,j=8.0hz,2h),7.43~7.32(m,2h),6.78(s,1h),6.13(dd,j1=2.0hz,j2=16.8hz,1h),5.70(d,j=10.4hz,1h),3.614(brs,8h);13cnmr(dmso-d6,400mhz)δ(ppm)169.32,164.39,137.04,134.04,133.88,128.05,127.67,125.89;ei/mass(m/z):calculated288.11,found[m+na]+310.9;ir(cm-1):3400,2926.0,2866.3,1606.5,1513.3,1438.8,1364.2,1006.4,849.8,790.2;dsc熔点测定发现其没有熔化,提示其可能是一个无定型的物质。实施例22.apcfpba的合成方法(1)具体步骤如下所示:将化合物1boc-哌嗪(57.0g,306mmol)和et3n(34.0g,336mmol,46.8ml)溶解在500mlch2cl2中,0℃条件下,将化合物1a丙烯酰氯(30.4g,336mmol,27.4ml)滴加到上述溶液中,混合液保持在15℃反应12h。液质监测反应完全后,反应液用水洗(300ml水,分别洗涤2次),水层用二氯甲烷萃取三次(50ml*3),保留有机层,合并后的有机层用无水硫酸钠干燥,旋去溶剂得到棕色油2(80.0g,粗品,1hnmr(cdcl3,400mhz)δ6.52(q,j=6.4hz,1h),6.26(dd,j1=1.6hz,j2=16.8hz,1h),5.70~5.62(m,1h),3.61~3.32(m,8h),1.43(s,9h))。化合物2(15.0g,62.4mmol)溶解在ch2cl2(100ml)中,然后加入tfa(21.4g,187mmol,13.9ml)。混合物在25℃条件下搅拌16小时。液质检测其反应基本完全,混合物浓缩后得到化合物3(18.0gcrude,tfasalt),是一个黄色的油。将1.45g的3a(7.87mmol)溶解在20ml的dmf中,加入4.49ghatu(11.8mmol)和2.39get3n(23.6mmol,3.29ml).混合物搅拌30min,然后化合物3(4.00g,15.7mmol,tfasalt)加入.混合物在25℃条件下搅拌16小时。lc-ms显示基本反应完全,混合物减压,用硅胶柱纯化,展开剂是(ch2cl2∶meoh=10∶1),再用制备液相纯化,(column:ymctriartc18,250*50mm,7μm;mobilephase:[water(0.1%tfa)-acn];b%:0%-32%,18min)得到目标产物(1.10g,45.7%yield),形态为白色固体。(2)apcfpba的pka测定称取适量的apcfpba或其类似物溶解于40ml的0.01n的naoh(150mmnacl)溶液中,用0.01n的hcl溶液在pka/logp测定仪进行检测,得到pka,已得到的apcfpba化合物的pka值为6.82。(3)结构信息合成的单体化合物经结构表征,包括核磁氢谱、碳谱、质谱、红外、dsc熔点检测,如下所示:1hnmr(dmso-d6,400mhz)δ(ppm)δ8.35(s,2h),7.67(d,j=7.6hz,1h),7.59(d,j=10.0hz,1h),7.41-7.37(m,1h),6.85-6.69(m,1h),6.15-6.11(m,1h),5.70(d,j=7.6hz,1h),3.66(s,4h),3.51(s,2h),3.23(s,2h):ei/mass(m/z):calculated306.1,found[m+h]+307.0;dsc熔点测定发现其超过300摄氏度没有熔化,提示其可能是一个无定型的物质;实施例33.apccf3pba的合成方法步骤如下所示:(1)boc-哌嗪丙烯酰胺的合成将化合物1,boc-哌嗪(57.0g,306mmol)和et3n(34.0g,336mmol,46.8ml)溶解在500mlch2cl2中,0℃条件下,将化合物1a丙烯酰氯(30.4g,336mmol,27.4ml)滴加到上述溶液中,混合液保持在15℃反应12h。液质监测反应完全后,反应液用水洗(300ml水,分别洗涤2次),水层用二氯甲烷萃取三次(50ml*3),保留有机层,合并后的有机层用无水硫酸钠干燥,旋去溶剂得到棕色油2(80.0g,粗品,1hnmr(cdcl3,400mhz)δ6.52(q,j=6.4hz,1h),6.26(dd,j1=1.6hz,j2=16.8hz,1h),5.70~5.62(m,1h),3.61~3.32(m,8h),1.43(s,9h))。(2)哌嗪丙烯酰胺的合成化合物2(15.0g,62.4mmol)溶解在ch2cl2(100ml)中,然后加入tfa(21.4g,187mmol,13.9ml)。混合物在25℃条件下搅拌16小时。液质检测其反应基本完全,混合物浓缩后得到化合物3(18.0gcrude,tfasalt),是一个黄色的油。(3)目标分子的合成化合物3a(900mg,3.85mmol)溶解在dmf(10.0ml),加入hatu(2.19g,5.77mmol)和et3n(1.17g,11.5mmol,1.61ml),混合物活化30min,然后将化合物3(2.93g,11.5mmol,tfasalt)加进去,混合物在25℃条件下搅拌16h,lcms显示已基本反应完全,混合物减压浓缩,再用硅胶柱预纯化(ch2cl2∶meoh=10∶1),再用制备色谱进行纯化得到apccf3白色粉末。(column:phenomenexlunac18250*50mm*10um;流动相:[water(0.1%tfa)-acn];b%:10%-40%,20min)。(4)apccf3pba及其类似物的pka测定称取适量的apccf3pba或其类似物溶解于40ml的0.01n的naoh(150mmnacl)溶液中,用0.01n的hcl溶液在pka/logp测定仪进行检测,得到pka,已得到的apccf3pba化合物的pka值为6.71。(5)结构信息:合成的单体化合物经结构表征,介于前面apcpba的结构表征,衍生物的结构确证减少部分分析,只检测包括核磁氢谱、质谱,检测得到apccf3pba的结构信息如下所示:1hnmr(dmso-d6,400mhz)δ(ppm)δ8.47(s,2h),8.17(d,j=17.2hz,1h),8.10(d,j=7.6hz,1h),7.55-7.44(m,1h),6.86-6.67(m,1h),6.14-6.10(m,1h),5.80-5.60(m,1h),3.73-3.61(m,6h),3.16-2.99(m,2h);ei/mass(m/z):calculated356.11,found[m+na]+357.10;dsc熔点测定发现其熔点范围为265.3-267.5℃。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1