一种回收利用醋酸的对位酯合成工艺的制作方法
本发明属于对位酯合成工艺技术领域,更具体地,涉及一种回收利用醋酸的对位酯合成工艺。
背景技术:
对位酯,也称“对氨基苯基-β-羟乙基砜硫酸酯”,是生产活性染料的重要中间体,主要用于制造km或m系列活性染料料。适用于活性翠蓝(kn-g)、活性黑(kn-b)、活性嫩黄(m-5g)、活性金黄(km-g)、活性艳红(km-2b、km-8b)等品种。近年来,我国乙烯砜型活性染料的生产比例逐年大幅上升,已发展成为活性染料中产量最大的一类。在活性染料中,对位酯既具有作为重氮化组分的氨基,又有可发生化学反应生成染料-纤维共价键的反应性基团,故能大大提高染料的利用率,其经济性也得到了大幅度提高。
目前对位酯的合成方法主要有三类:1、乙酰苯胺路线,2、对硝基氯苯路线,3、硝基苯路线。在以上合成路线中,硝基苯路线收率不高,工业价值不大。对硝基氯苯路线成本低,但目前处于技术探索阶段。乙酰苯胺路线是目前生产对位酯的主要路线。
因此,在对位酯的生产过程中,氯磺化工艺往往必不可少,而在氯磺化工艺中,通常会用到乙酰苯胺(又称“退热冰”),实际生产时,所需的乙酰苯胺大多通过采购来获取,导致对位酯生产过程中的原材料成本较高;此外,在现有对位酯的制备工艺中,酯化反应往往必不可少,在酯化反应过程中,通常会产生醋酸,而现有的对位酯合成工艺中,大多没有将醋酸进行合理的回收利用,或者直接将低浓度醋酸廉价出售,吸收过程中造成含有醋酸的废气排放到空气中,无法实现醋酸的有效回收和循环利用,不仅造成资源的浪费,还会造成环境污染,无法实现对位酯生产过程中的节能环保。
技术实现要素:
针对现有技术的以上缺陷或改进需求,本发明提供一种回收利用醋酸的对位酯合成工艺,回收酯化环节产生的醋酸,与苯胺反应后制备乙酰苯胺,作为氯磺化环节的原料,进而用于制备对位酯,实现了对位酯制备过程中产生的醋酸的有效利用。
为实现上述目的,提供一种回收利用醋酸的对位酯合成工艺,其特征在于,包括如下步骤:
(1)制备乙酰苯胺
回收酯化反应中产生的83%~87%的醋酸,进入蒸馏装置分馏出高纯度99.5%以上的醋酸,将高纯度的所述醋酸加入到乙酰化一级合成釜,再将在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;所述气液相反应后的液体溢流到乙酰化四级合成釜进行反应,将所述乙酰化四级合成混合液送入蒸馏釜进行减压蒸馏,将过量的醋酸、未反应的苯胺以及少量的水蒸出,重新送回反应装置反应制备所述乙酰苯胺;所述减压蒸馏后的熔融物为乙酰苯胺,通过切片制成成品用于对位酯的氯磺化环节中;
(2)制备对位酯
向所述磺化反应釜中加入设定量的氯磺酸和所述乙酰苯胺,反应得到磺化物,并添加氯化亚砜,反应制备得到氯磺化物;稀释抽滤后进行还原反应、缩合反应及酯化反应,最后将酯化反应后的产物粉碎包装,制备得到所述对位酯。
进一步地,减压蒸馏后的所述醋酸的含量为40%~50%。
进一步地,所述减压蒸馏控制在三米蒸馏塔顶温度225-248℃,真空泵达到335-350mmhg,蒸出物颜色由淡黄色变白,流速减缓时即为达到蒸馏终点,停止减压开始压料。
进一步地,所述成品乙酰苯胺为片状。
进一步地,所述乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.45~0.46:1:0.46~0.47。
进一步地,所述氯磺化物的制备包括如下步骤:
向低温磺化锅中加入设定的氯磺酸和乙酰苯胺反应得到磺化物;投料温度控制在25-35℃,投料时间为4~5小时;投料完成后,维持4~5小时,温度控制在25~35℃;
将所述低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;所述高温磺化的温度为45~48℃,维持1~2小时;
加入氯化亚砜进行反应,时间控制在4~5小时,所述氯化亚砜添加完后,维持温度在55~60℃并保持2~3小时,再降温至40~50℃放料。
进一步地,所述缩合反应包括如下步骤:
将所述还原液打入缩合反应装置中,抽真空,关闭真空阀,充入氮气,锅内压力为正即可,并升温至55~60℃,投入设定量的磷酸钠;
加入环氧乙烷,控制反应温度在45~50℃,加入环氧乙烷的时间控制在3~4小时;
所述环氧乙烷添加完毕后,升温至50℃维持3~4小时,维持完毕取样分析,合格后开循环水降温至25~30℃时放料。
进一步地,还原液:环氧乙烷:磷酸钠的摩尔比,1:0.52~0.53:0.0014。
进一步地,所述酯化反应中,包括如下步骤:向所述缩合物中缓慢加入硫酸,200-210℃下搅拌8~16分钟,当物料将成干粉时,开始计时维持2~3小时后放料;
进一步地,所述缩合物与硫酸的摩尔比为1:1.03~1.06。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果:
(1)本发明的回收利用醋酸的对位酯合成工艺,回收酯化环节产生的醋酸,与苯胺反应后制备乙酰苯胺,作为氯磺化环节的原料,进而用于制备对位酯,实现了对位酯制备过程中产生的醋酸的有效利用,减少了醋酸向大气的排放。
(2)本发明的回收利用醋酸的对位酯合成工艺,减压蒸馏后的过量的醋酸、未反应的苯胺以及少量的水重新送回反应装置反应制备乙酰苯胺,进一步对其进行了循环利用。
(3)本发明的回收利用醋酸的对位酯合成工艺,优化了乙酰苯胺、氯磺酸、氯化亚砜的摩尔比,相比现有技术中对于三者的配比,减少了氯磺酸的用量,用氯化亚砜替换部分的氯磺酸,通过配比的优化,使对位酯收率得以提升。
(4)本发明的回收利用醋酸的对位酯合成工艺,对位酯合成方法的缩合反应环节中,加入磷酸钠作为缓冲剂,稳定ph值,抑制环氧乙烷的水解,从而减少环氧乙烷的使用量,降低成本。且通过调整还原产物、环氧乙烷及磷酸盐的质量摩尔比,进一步提高了收率。
(5)本发明的回收利用醋酸的对位酯合成工艺,在氯磺酸基础上添加氯化亚砜,提高了收率(原75%提高到82%以上),使磺化反应的速率加快,反应平稳,磺化反应效率高,副反应少,环境效果好,以有效的提高磺化反应的收率,减少副广物的产生,进而减少了废水排放量,降低材料消耗,提高产品质量。
附图说明
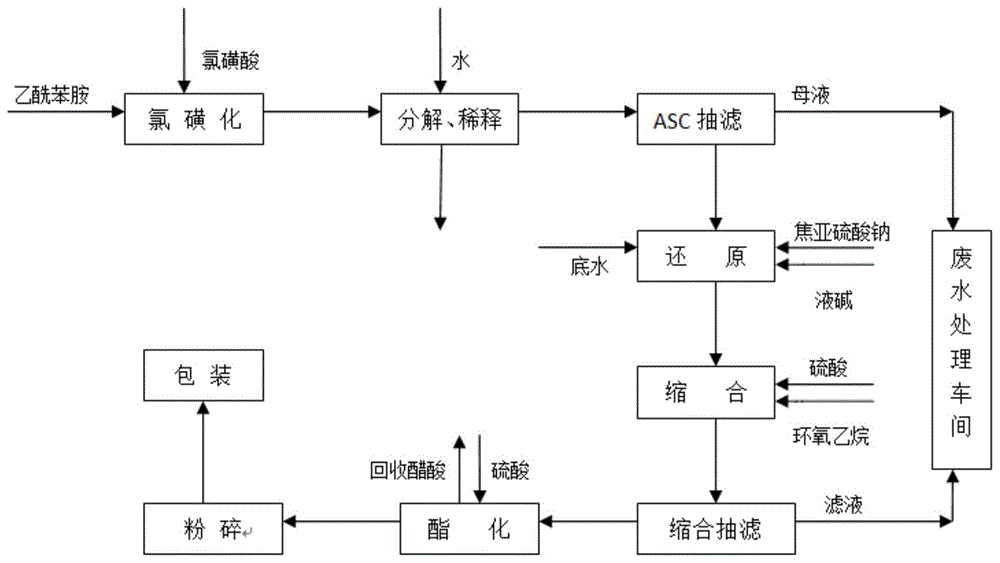
图1为本发明实施例一种回收利用醋酸的对位酯合成工艺流程图;
图2为本发明实施例一种回收利用醋酸的对位酯合成工艺涉及的乙酰苯胺合成环节流程图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
图1为本发明实施例一种回收利用醋酸的对位酯合成工艺流程图。图2为本发明实施例一种回收利用醋酸的对位酯合成工艺涉及的乙酰苯胺合成环节流程图。结合图1和图2,本发明的回收利用醋酸的对位酯合成工艺包括如下步骤:
(1)制备乙酰苯胺
s11:回收酯化反应中产生的83%~87%的醋酸,进入蒸馏装置分馏出高纯度99.5%以上的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中进行减压蒸馏,当三米蒸馏塔顶温度225-248℃,真空泵达到335-350mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、未反应的苯胺及少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
其中,s12中,需控制在三米蒸馏塔顶温度225-248℃,真空泵达到335-350mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,因为在此温度范围内,可以将水和醋酸一起蒸馏出来。且此时蒸发出的醋酸含量在40%~50%,俗称真空酸。s14中,优选地,熔融的乙酰苯胺,可通过压缩空气将其压到保温罐中进行保温处理,继而输送进滚筒制片机中制成片状的乙酰苯胺,得到片状退热冰待用。
本发明的回收利用醋酸的对位酯合成工艺,回收酯化环节产生的醋酸,与苯胺反应后制备乙酰苯胺,作为氯磺化环节的原料,进而用于制备对位酯,实现了对位酯制备过程中产生的醋酸的有效利用,减少了醋酸向大气的排放。
本发明的回收利用醋酸的对位酯合成工艺,减压蒸馏后的过量的醋酸、未反应的苯胺以及少量的水重新送回反应装置反应制备乙酰苯胺,进一步对其进行了循环利用。
(2)制备对位酯
s21:向磺化反应釜中加入设定量的氯磺酸和乙酰苯胺,反应得到磺化物,并添加氯化亚砜,反应制备得到氯磺化物;
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.45~0.46:1:0.46~0.47;
s22:将氯磺化物进行稀释后抽滤;
s23:加入焦亚硝酸盐,并调节稳定的ph值,进行还原反应得到还原液;
s24:以磷酸钠作为缓冲剂,加入硫酸和环氧乙烷,进行缩合反应得到缩合产物;
s25:将缩合产物进行抽滤;
s26:在抽滤后的产物中加入硫酸,进行酯化反应;
s27:将酯化反应后的产物粉碎包装,制备得到对位酯。
具体地,步骤s21中,包括如下步骤:
s211:向低温磺化锅中加入设定的氯磺酸和乙酰苯胺反应得到磺化物;投料温度控制在25-35℃,投料时间为4~5小时;投料完成后,维持4~5小时,温度控制在25~35℃;
s212:将所述低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;所述高温磺化的温度为45~48℃,维持1~2小时;
s213:加入氯化亚砜进行反应。加入氯化亚砜的时间控制在4~5小时,所述氯化亚砜添加完后,维持温度在55~60℃并保持2~3小时,再降温至40~50℃放料。
步骤s23中,向氯磺化物中加入焦亚硫酸钠,搅拌至焦亚硫酸钠完全溶解后,采用碱液调整ph值保持碱性,优选为7.0~7.5,控制反应温度,优选为31~35℃,反应完毕后,将其转移至缩合反应釜中,进行下一步反应。
进一步地,步骤s24中,具体包括如下步骤:
s241:将所述还原液打入缩合反应装置中,抽真空,并升温,投入设定量的磷酸钠;
s242:加入环氧乙烷,控制反应温度在45~50℃,加入环氧乙烷的时间控制在3~4小时;
s243:所述环氧乙烷添加完毕后,升温至50℃维持3~4小时,维持完毕取样分析,合格后开循环水降温至25~30℃时放料。
其中,还原液:环氧乙烷:磷酸钠的摩尔比为1:0.52~0.53:0.0014,更为优选地,还原液:环氧乙烷:磷酸钠的摩尔比为1:0.525:0.0014。步骤s42中,加环氧乙烷过程中控制ph值在7.5~8之间,步骤s43中,维持过程中,ph始终维持在碱性环境。步骤s24中缩合后的产物主要为对-β-羟乙基砜基乙酰苯胺。
本发明对位酯合成方法的缩合反应环节中,加入磷酸钠作为缓冲剂,稳定ph值,抑制环氧乙烷的水解,从而减少环氧乙烷的使用量,降低成本。且通过调整还原产物、环氧乙烷及磷酸盐的摩尔比,从现有的比例1:0.57:0.00.14调整至1:0.52~0.53:0.0014,进一步提高了收率。
进一步地,步骤s26中,向缩合物中缓慢加入硫酸,200-210℃下搅拌8~16分钟,当物料将成干粉时,开始计时维持2~3小时后放料。所述缩合物与硫酸的摩尔比为1:1.03~1.06。所述硫酸为100%硫酸。对位酯合成方法的酯化环节中,提高了硫酸的浓度,将现有技术中常用的98%的浓硫酸优化为100%浓硫酸,进而提升了质量和收率。
本发明对位酯合成方法中,在氯磺酸基础上添加氯化亚砜,提高了收率(原75%提高到82%以上),使磺化反应的速率加快,反应平稳,磺化反应效率高,副反应少,环境效果好,以有效的提高磺化反应的收率,减少副广物的产生,进而减少了废水排放量,降低材料消耗,提高产品质量。优化了乙酰苯胺、氯磺酸、氯化亚砜的摩尔比,相比现有技术中对于三者的配比,减少了氯磺酸的用量,用氯化亚砜替换部分的氯磺酸,通过配比的优化,使对位酯收率提高了5%左右。
本更好的解释本发明的对位酯合成方法,提供如下具体实施例:
实施例1
(1)制备乙酰苯胺
s11:回收酯化反应中产生的85%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度225℃,真空泵达到335mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺45mol反应;投料温度控制在25℃,投料时间为4小时;投料完成后,维持4小时,温度控制在25℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为45℃,维持1小时;
加入46mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4小时,氯化亚砜添加完后,维持温度在55℃并保持2小时,再降温至40℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.45:1:0.46。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至55℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.5,不允许偏酸或偏碱。温度控制在45℃。加环氧时间为3小时;
环氧乙烷加完后,升温50℃维持3小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至25℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.52:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在200℃,搅拌8分钟,再缓慢加入103mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.03。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例1制备得到的对位酯收率进行测定,得到本实施例的收率为82%。
实施例2
(1)制备乙酰苯胺
s11:回收酯化反应中产生的83%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度230℃,真空泵达到340mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺45mol反应;投料温度控制在26℃,投料时间为4.2小时;投料完成后,维持4小时,温度控制在27℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为45℃,维持1小时;
加入47mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.4小时,氯化亚砜添加完后,维持温度在55℃并保持2小时,再降温至40℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.45:1:0.47。
s22:加入焦亚硝酸盐,用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至55℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52.1mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.7,不允许偏酸或偏碱。温度控制在45℃。加环氧时间为3小时;
环氧乙烷加完后,升温50℃维持3小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至25℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.521:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在200℃,搅拌10分钟,再缓慢加入104mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.04。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例2制备得到的对位酯收率进行测定,得到本实施例的收率为83%。
实施例3
(1)制备乙酰苯胺
s11:回收酯化反应中产生的87%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度235℃,真空泵达到345mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺46mol反应;投料温度控制在28℃,投料时间为4.4小时;投料完成后,维持5小时,温度控制在29℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为45℃,维持1小时;
加入46mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.4小时,氯化亚砜添加完后,维持温度在57℃并保持2小时,再降温至40℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.46:1:0.46。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至55℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52.2mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.8,不允许偏酸或偏碱。温度控制在45℃。加环氧时间为3小时;
环氧乙烷加完后,升温50℃维持3小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至25℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.522:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在200℃,搅拌10分钟,再缓慢加入104mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.04。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例3制备得到的对位酯收率进行测定,得到本实施例的收率为85%。
实施例4
(1)制备乙酰苯胺
s11:回收酯化反应中产生的85%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度248℃,真空泵达到350mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺46mol反应;投料温度控制在30℃,投料时间为4.6小时;投料完成后,维持4小时,温度控制在30℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为46℃,维持1小时;
加入47mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.4小时,氯化亚砜添加完后,维持温度在60℃并保持2小时,再降温至40℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.46:1:0.47。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至55℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52.3mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在8.0,不允许偏酸或偏碱。温度控制在45℃。加环氧时间为3小时;
环氧乙烷加完后,升温50℃维持4小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至28℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.523:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在210℃,搅拌10分钟,再缓慢加入105mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.05。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例4制备得到的对位酯收率进行测定,得到本实施例的收率为87%。
实施例5
(1)制备乙酰苯胺
s11:回收酯化反应中产生的85%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度240℃,真空泵达到340mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺45.5mol反应;投料温度控制在30℃,投料时间为4.6小时;投料完成后,维持4小时,温度控制在30℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为46℃,维持1小时;
加入46mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.4小时,氯化亚砜添加完后,维持温度在60℃并保持2小时,再降温至40℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.455:1:0.46。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至55℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52.4mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.9,不允许偏酸或偏碱。温度控制在45℃。加环氧时间为3小时;
环氧乙烷加完后,升温50℃维持4小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至28℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.524:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在210℃,搅拌12分钟,再缓慢加入106mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.06。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例5制备得到的对位酯收率进行测定,得到本实施例的收率为85%。
实施例6
(1)制备乙酰苯胺
s11:回收酯化反应中产生的85%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度240℃,真空泵达到340mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺45.5mol反应;投料温度控制在30℃,投料时间为4.6小时;投料完成后,维持4小时,温度控制在30℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为46℃,维持1小时;
加入47mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.6小时,氯化亚砜添加完后,维持温度在57℃并保持3小时,再降温至42℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.455:1:0.47。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至55℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52.6mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.8,不允许偏酸或偏碱。温度控制在50℃。加环氧时间为4小时;
环氧乙烷加完后,升温50℃维持4小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至28℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.526:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在210℃,搅拌14分钟,再缓慢加入106mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.06。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例6制备得到的对位酯收率进行测定,得到本实施例的收率为83%。
实施例7
(1)制备乙酰苯胺
s11:回收酯化反应中产生的85%醋酸,进入蒸馏装置分馏出高纯度99.6%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度238℃,真空泵达到350mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺45mol反应;投料温度控制在35℃,投料时间为4.8小时;投料完成后,维持5小时,温度控制在32℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为48℃,维持1小时;
加入46.5mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.6小时,氯化亚砜添加完后,维持温度在60℃并保持3小时,再降温至45℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.45:1:0.465。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至60℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷52.8mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.8,不允许偏酸或偏碱。温度控制在50℃。加环氧时间为4小时;
环氧乙烷加完后,升温50℃维持3小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至28℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.528:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在210℃,搅拌16分钟,再缓慢加入105mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.05。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例7制备得到的对位酯收率进行测定,得到本实施例的收率为83%。
实施例8
(1)制备乙酰苯胺
s11:回收酯化反应中产生的85%醋酸,进入蒸馏装置分馏出高纯度99.5%的醋酸,高纯度的醋酸加入到乙酰化一级合成釜,然后再把在乙酰化合成中分馏出的苯胺与原料苯胺按0.3:1的比例加入乙酰化三级合成釜,进行气液相反应;气液相反应的液体溢流到乙酰化四级合成釜进行反应。
s12:将混合液间断加入蒸馏罐中,三米蒸馏塔顶温度240℃,真空泵达到337mmhg,蒸出物颜色由淡黄色变白,流速有些慢时即为达到蒸馏终点,停止减压开始压料,将过量的醋酸、少量的水蒸出,其中,减压蒸馏后的熔融物为乙酰苯胺。
s13:过量的醋酸、少量的水重新送回反应装置反应制备乙酰苯胺;
s14:将熔融的乙酰苯胺经过保温后制成成品乙酰苯胺用于对位酯的氯磺化环节中。
(2)制备对位酯
s21:向低温磺化锅中加入设定的氯磺酸100mol和乙酰苯胺46mol反应;投料温度控制在35℃,投料时间为5小时;投料完成后,维持5小时,温度控制在35℃;
将低温磺化锅中制备得到的所述磺化物转入高温磺化锅中进行高温磺化;高温磺化的温度为48℃,维持1小时;
加入46.5mol氯化亚砜进行反应,加入氯化亚砜的时间控制在4.6小时,氯化亚砜添加完后,维持温度在60℃并保持2小时,再降温至50℃放料,将磺化物进行稀释后抽滤。
乙酰苯胺、氯磺酸及氯化亚砜的摩尔比为0.46:1:0.465。
s22:加入焦亚硝酸盐,并用液碱调节稳定的ph值7.5,进行还原反应得到还原液;
s23:将100mol高温还原液打入缩合锅中,封盖,抽真空(负压为-0.01mpa即可),关闭真空阀,充入氮气,锅内压力为正即可,然后升温至60℃,启动物料循环泵投入0.14mol的磷酸钠;
用氮气置换一下环氧乙烷进料管及缩合釜,防止形成可爆炸性混合气体。开始加入环氧乙烷53mol,同时打开稀酸进料阀,加环氧乙烷过程中控制ph值在7.8,不允许偏酸或偏碱。温度控制在50℃。加环氧时间为4小时;
环氧乙烷加完后,升温50℃维持3小时,维持过程中ph值始终维持在碱性环境。维持完毕取样分析,合格后开循环水降温至28℃时放料。
还原液:环氧乙烷:磷酸钠的摩尔比为1:0.53:0.0014。
s24:将缩合产物进行抽滤,在抽滤后的产物中加入硫酸,进行酯化反应。投入100mol的缩合离心物料,干粉进完,同时打开油阀进油,油温维持在210℃,搅拌10分钟,再缓慢加入105mol的100%酸,当物料将成干粉时,开始计时维持2小时,然后取样,合格后放料;
缩合物与缩合物:100%硫酸的摩尔比为1:1.05。
s25:将酯化反应后的产物粉碎包装,制备得到对位酯。
将实施例8制备得到的对位酯收率进行测定,得到本实施例的收率为83%。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!