不同顺反异构体比例维生素K1及其中间体卤代植物醇的制备方法与流程
本发明属于药物化学领域,具体涉及一种不同顺反异构体比例维生素k1及其中间体卤代植物醇的制备方法。
背景技术:
维生素k1(vitamink1),化学名为2-甲基-3-(3,7,11,15-四甲基十六-2-烯基)-1,4-萘醌,属于维生素类药物,分子侧链上有一个双键,因而存在顺反异构体。化学合成维生素k1为顺式和反式异构体的混合物,而顺式异构体几乎没有生理活性。因此20世纪80年代以后,各国药典均对维生素k1的顺式异构体规定了限量(顺式异构体≤21.0%)。
目前,国内维生素k1制备方法主要有1)傅克烷基化法;2)甲萘醌-环戊二烯法。傅克烷基化法的缺点是产率低,产品杂质多,纯化困难;而甲萘醌-环戊二烯法步骤操作简单,产品收率高,产品纯化难度较低,具有工业生产价值。
季亚飞等(ya-feiji,etal.improvedsynthesisofvitamink1,syntheticcommunica-tions,vol.33,no.5,pp.763-772,2003)报道以异植醇为原料合成维生素k1,异植物醇与氯化氢溶液反应得到的氯代植醇产物顺反比例为19.5:80.5,与溴化氢溶液合成得到的溴代植醇产物顺反比例为24.5:75.5;与三溴化磷反应得到的溴代植醇产物顺反例为30:70;以天然植物醇为原料,与三溴化磷合成得到的溴代植醇产物顺反比例在10:85~15:90。因为维生素k1中顺反异构体的比例与卤代植物醇保持一致,因此该方法得到的顺式异构体比例均为10%以上。
张志刚等(不同顺反异构体比例的维生素k1的合成.中国医药工业杂志,47(2),pp.147-148,2016.)报道以天然植物醇在不同量的水介质作用下,经三溴化磷溴化,可得到不同顺反异构体比例的叶基溴,然后再与环戊二烯甲萘醌缩合得到维生素k1前体,最后解聚得到顺式异构体比例为10~21%或≤1%的维生素k1。
因此现有技术这些方法通常难以控制产物维生素k1中顺式异构体的比例在1~21%这个较大的范围内可调控,特别是难以得到顺式异构体的比例为1~10%的维生素k1。
因此,本领域仍需开发操作简便、异构体比例可调的维生素k1制备方法。
技术实现要素:
本发明的目的是提供了一种不同顺反异构体比例维生素k1及其中间体卤代植物醇的制备方法,解决现有技术中存在的异构体比例不可控的问题。
本发明的目的之一采用以下方案:
一种不同顺反异构体比例维生素k1中间体卤代植物醇的制备方法,通过将植物醇在有机溶剂和磷酸条件下与三卤化磷在一定温度条件下进行卤代反应,得到卤代植物醇,其反应如下所示:
作为本发明的进一步改进,所述有机溶剂为饱和脂肪烃或醚类溶剂中的其中一种。
作为本发明的进一步改进,所述饱和脂肪烃优选自正戊烷、正己烷、正庚烷、石油醚中的其中一种;所述醚类溶剂优选为乙醚、四氢呋喃中的其中一种,所选用的有机溶剂后处理简单,价格低廉;其中更优选饱和脂肪烃溶剂,如正己烷其对植物醇、卤代植物醇和维生素k1的溶解性更好,能够有效降低反应中的杂质。
作为本发明的进一步改进,所述三卤化磷为三氯化磷或三溴化磷中的其中一种,更优选为三溴化磷,因为后续反应中作为起始原料的溴代植物醇较氯代植物醇活性更高。
作为本发明的进一步改进,所述卤代反应中磷酸与植物醇的质量比为0.05:1~0.6:1,当磷酸与植物醇的质量比小于0.05:1时,反应较慢,且杂质含量和杂质种类较多;进一步优选为0.2:1~0.5:1,当磷酸与植物醇的质量比介于0.2:1~0.5:1之间时,所得维生素k1产品纯度较高且杂质含量和杂质种类较少,顺式异构体比例低于21.0%;当磷酸与植物醇的质量比超过0.6:1时,顺式异构体比例超出21.0%,需要纯化去除顺式异构体,造成成本上升,且杂质含量和杂质种类明显增加。
作为本发明的进一步改进,所述卤代反应中反应温度为-20~20℃,进一步优选为-10~10℃,当温度超过10℃时,会产生相对较多的杂质。
本发明的目的之二,提供了一种不同顺反异构体比例维生素k1的制备方法,
1)将植物醇在有机溶剂和磷酸条件下与三卤化磷在一定温度下进行卤代反应,得到卤代植物醇;
2)卤代植物醇在叔丁醇钾和四氢呋喃条件下与环戊二烯甲萘醌进行缩合反应,降温至-20~20℃,得到维生素k1前体;
3)将得到的维生素k1前体用冰乙酸溶解,升温至90~110℃,进行解聚合反应得到维生素k1,其反应式如下所示:
作为本发明的进一步改进,所述步骤1)中有机溶剂为正戊烷、正己烷、正庚烷、石油醚、乙醚、四氢呋喃中的其中一种,所述的三卤化磷为三氯化磷或三溴化磷中的其中一种。
作为本发明的进一步改进,所述步骤1)中反应温度为-20~20℃,磷酸与植物醇的质量比为0.05:1~0.6:1。
本发明所述的原材料都是常规使用的,均可以从市场购得。
本发明采用液相色谱法检测产品维生素k1的顺式异构体比例。
本发明相对现有技术,具有以下优势:将植物醇在有机溶剂和磷酸条件下与三卤化磷在一定温度条件下反应,通过控制卤代反应中磷酸加入量,使得到的维生素k1顺式异构体比例在1%至21%之间这个较大的范围内可调控,特别是很容易得到顺式异构体的比例为1~10%的维生素k1。采用本发明所述的技术得到的维生素k1,纯度方面基本可以和现有技术水平相当,且异构体≤21.0%,符合各国药典标准。本发明技术克服了目前国内维生素k1生产在不加水条件下,顺式异构体比例只能控制在≤1%;在加不同量水介质作用下方法纯度较低、杂质较多,产品纯化难,顺式异构体不可控的缺点,本发明操作更简便,纯化难度更低,有利于工业化生产应用。
附图说明
图1是实施例1所得顺反异构体维生素k1的hplc图谱。
图2是实施例2所得顺反异构体维生素k1的hplc图谱。
图3是实施例3所得顺反异构体维生素k1的hplc图谱。
图4是实施例4所得顺反异构体维生素k1的hplc图谱。
图5是实施例5所得顺反异构体维生素k1的hplc图谱。
图6是实施例6所得顺反异构体维生素k1的hplc图谱。
图7是实施例7所得顺反异构体维生素k1的hplc图谱。
图8是实施例8所得顺反异构体维生素k1的hplc图谱。
图9是实施例9所得顺反异构体维生素k1的hplc图谱。
图10是实施例10所得顺反异构体维生素k1的hplc图谱。
图11是对比例1所得顺反异构体维生素k1的hplc图谱。
图12是对比例2所得顺反异构体维生素k1的hplc图谱。
具体实施方式
为便于本领域技术人员理解本发明内容,下面将结合具体实施例进一步描述本发明的技术方案,但以下内容不是针对本发明权利要求书请求保护的范围和精神做出限制。
实施例1
将200g植物醇、10g磷酸加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过3小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
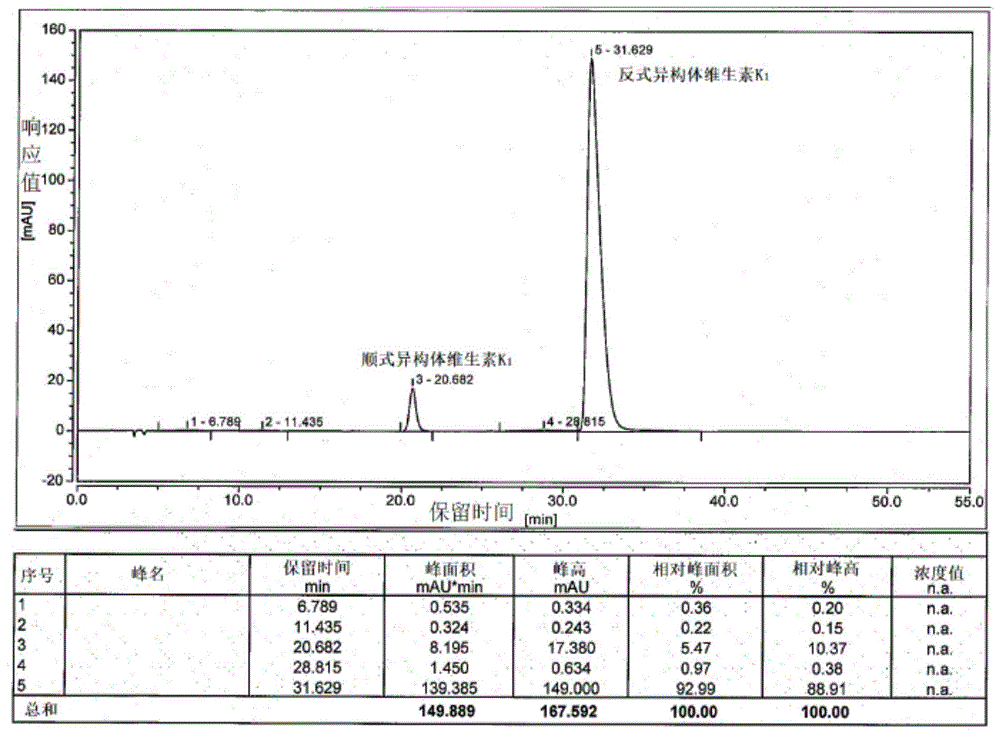
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图1所示,经hplc检测,保留时间为20.682min的峰为顺式异构体,其峰面积百分比为5.47%;保留时间为31.629min的峰为反式异构体,其峰面积百分比为92.99%;因此顺反异构体比例为5.6:94.4;最终产品顺反异构体的色谱纯度为98.46%,总杂含量为1.54%,包含3种杂质。
实施例2
将200g植物醇、40g磷酸加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过2.5小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图2所示,经hplc检测,保留时间为20.509min的峰为顺式异构体,其峰面积百分比为9.93%;保留时间为30.295min的峰为反式异构体,其峰面积百分比为89.98%;因此顺反异构体比例为9.9:90.1;最终产品顺反异构体的色谱纯度为99.91%,总杂含量为0.09%,仅包含1种杂质。
实施例3
将200g植物醇、60g磷酸加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过2小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图3所示,经hplc检测,保留时间为26.173min的峰为顺式异构体,其峰面积百分比为15.10%;保留时间为35.960min的峰为反式异构体,其峰面积百分比为84.85%;因此顺反异构体比例为15.1:84.9;最终产品顺反异构体的色谱纯度为99.95%,总杂含量为0.05%,仅包含1种杂质。
实施例4
将200g植物醇、100g磷酸加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过1小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图4所示,经hplc检测,保留时间为36.364min的峰为顺式异构体,其峰面积百分比为18.52%;保留时间为46.364min的峰为反式异构体,其峰面积百分比为81.42%;因此顺反异构体比例为18.5:81.5;最终产品顺反异构体的色谱纯度为99.94%,总杂含量为0.06%,仅包含1种杂质。
实施例5
将200g植物醇、120g磷酸加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc经过1小时后监测原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图5所示,经hplc检测,保留时间为20.449min的峰为顺式异构体,其峰面积百分比为20.33%;保留时间为30.055min的峰为反式异构体,其峰面积百分比为75.82%;因此顺反异构体比例为21.1:78.9;最终产品顺反异构体的色谱纯度为96.15%,总杂含量为3.85%,包含8种杂质。
通过上述实施例1~5的对比可知,在其它工艺参数完全一致的条件下,研究了磷酸的不同加入量对产品中顺反异构体比例、产品纯度以及杂质的影响,实施例1中当磷酸与植物醇的质量比小于0.05:1时,反应较慢,且杂质含量和杂质种类较多;实施例2~4中当磷酸与植物醇的质量比介于0.2:1~0.5:1之间时,所得维生素k1产品纯度较高且杂质含量和杂质种类较少,顺式异构体比例低于21.0%;实施例5中当磷酸与植物醇的质量比超过0.6:1时,顺式异构体比例超出21.0%,需要纯化去除顺式异构体,造成成本上升,且杂质含量和杂质种类明显增加。
实施例6
将200g植物醇、60g磷酸加入1.2l正己烷中搅拌,降温至10~20℃,滴加240g三溴化磷,控温内温不高于20℃。tlc监测经过1小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至0~10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过1小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图6所示,经hplc检测,保留时间为23.792min的峰为顺式异构体,其峰面积百分比为13.65%;保留时间为36.605min的峰为反式异构体,其峰面积百分比为83.41%;因此顺反异构体比例为14.0:86.0;最终产品顺反异构体的色谱纯度为97.06%,总杂含量为2.94%,包含5种杂质。
通过对比实施例3和实施例6可知,在其它工艺参数完全一致的条件下,研究了反应温度对产品中顺反异构体比例、产品纯度以及杂质的影响,当溴代反应和缩合反应的温度不超过10℃时,会产生相对较少的杂质含量和种类,产品纯度更高。
实施例7
将200g植物醇、60g磷酸加入1.2l正己烷中搅拌,降温至-10~0℃,滴加130g三氯化磷,控温内温不高于10℃。tlc监测经过2小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得氯代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加氯代植物醇。tlc监测经过6小时后原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图7所示,经hplc检测,保留时间为20.775min的峰为顺式异构体,其峰面积百分比为15.13%;保留时间为31.201min的峰为反式异构体,其峰面积百分比为84.25%;因此顺反异构体比例为15.2:84.8;最终产品顺反异构体的色谱纯度为99.38%,总杂含量为0.62%,仅包含2种杂质。
通过对比实施例3和实施例7可知,在其它工艺参数完全一致的条件下,研究了三卤化磷中取代基的种类对反应的影响,结果表明:后续缩合反应中作为起始原料的溴代植物醇比氯代植物醇反应活性更高,反应时间更短;最终产品顺反异构体比例、产品纯度以及杂质含量相当,影响不大。
实施例8
将200g植物醇、100g磷酸加入1.2l四氢呋喃中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过1小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l四氢呋喃溶解,水洗,旋干有机相得维生素k1。如图8所示,经hplc检测,保留时间为22.522min的峰为顺式异构体,其峰面积百分比为17.18%;保留时间为35.162min的峰为反式异构体,其峰面积百分比为81.73%;因此顺反异构体比例为17.4:82.6;最终产品顺反异构体的色谱纯度为98.91%,总杂含量为1.09%,包含8种杂质。
通过对比实施例4和实施例8可知,在其它工艺参数完全一致的条件下,研究了卤代反应溶剂的种类对反应的影响,结果表明:正己烷对植物醇、卤代植物醇和维生素k1的溶解性更好,能够有效降低反应中的杂质含量和种类,产品纯度更高。
实施例9
将200g植物醇、30g磷酸加入1.2l正戊烷中搅拌,降温至-5~5℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过2.5小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正戊烷溶解,水洗,旋干有机相得维生素k1。如图9所示,经hplc检测,保留时间为24.886min的峰为顺式异构体,其峰面积百分比为7.16%;保留时间为37.452min的峰为反式异构体,其峰面积百分比为92.34%;因此顺反异构体比例为7.2:92.8;最终产品顺反异构体的色谱纯度为99.50%,总杂含量为0.5%,包含10种杂质。
实施例10
将200g植物醇、40g磷酸加入1.2l正庚烷中搅拌,降温至-5~5℃,滴加240g三溴化磷,控温内温不高于10℃。tlc监测经过3小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正庚烷溶解,水洗,旋干有机相得维生素k1。如图10所示,经hplc检测,保留时间为22.478min的峰为顺式异构体,其峰面积百分比为11.63%;保留时间为34.885min的峰为反式异构体,其峰面积百分比为87.88%;因此顺反异构体比例为11.7:88.3;最终产品顺反异构体的色谱纯度为99.51%,总杂含量为0.49%,包含7种杂质。
通过对比实施例2、实施例9和实施例10可知,结果表明卤代反应中脂肪烃溶剂种类变化、磷酸投料量的微小变化(控制磷酸与植物醇的质量比在0.2:1~0.5:1范围内变化)、反应温度的微弱升高对产品的纯度影响不是很明显,但会导致最终产品的杂质种类明显增加,可能是因为杂质种类对温度相对较敏感。
对比例1
将200g植物醇加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过4小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图11所示,经hplc检测,保留时间为20.714min的峰为顺式异构体,其峰面积百分比为1.13%;保留时间为30.254min的峰为反式异构体,其峰面积百分比为98.43%;因此顺反异构体比例为1.1:98.9。当反应中不加磷酸或水时,顺式异构体比例始终很低,接近1%含量。
对比例2
将200g植物醇、6g纯化水加入1.2l正己烷中搅拌,降温至-10~0℃,滴加240g三溴化磷,控温内温不高于0℃。tlc监测经过4小时后原料反应完全,加入甲醇淬灭,分层,旋干去除溶剂,得溴代植物醇。
将11.5g叔丁醇钾和1.2l四氢呋喃加入反应瓶中搅拌,降温至-20~-10℃,加入60g环戊二烯甲萘醌,缓慢滴加溴代植物醇。tlc监测经过2小时原料反应完全,加入盐酸淬灭,旋蒸除去四氢呋喃,剩余水相用0.5l甲基叔丁基醚萃取,分液,旋蒸去除溶剂,得油状物。油状物用0.5l冰乙酸溶解,加入反应瓶中搅拌,升温至95~100℃,tlc监测原料反应完全,旋干,加入0.5l正己烷溶解,水洗,旋干有机相得维生素k1。如图12所示,经hplc检测,保留时间为24.392min的峰为顺式异构体,其峰面积百分比为5.54%;保留时间为38.225min的峰为反式异构体,其峰面积百分比为86.29%;因此顺反异构体比例为6.0:94.0。同时在其它组成投料量不变时,控制纯化水的不同加入量,如纯化水加入量分别为1g或4g,该方法得到的顺反异构体比例分别为4.3:95.7和5.9:94.1,即顺式异构体的比例在6%以下,无法做到报道所述的异构体比例。
以上显示和描述了本发明的基本原理和主要特征以及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都要落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!