引物探针组合、试剂盒及其用于检测ACTN3基因突变的应用的制作方法
本发明涉及生物技术领域,特别涉及引物探针组合、试剂盒及其用于检测actn3基因突变的应用。
背景技术:
人类基因组中最常见的基因突变是snp。人类基因组中总共有300万个snp,平均每1000个碱基中就有一个snp,所以snp是人类基因组中密度最大的遗传标记。snp发生频率较高,被认为是继微卫星之后的新一代遗传学标记,在医学遗传学、药物遗传学、疾病遗传学、疾病诊断学以及人类进化等研究领域都有着很高的研究价值和应用前景。actn3基因的snps与人体运动能力潜在相关,actn3r77x是actn3基因位于rs1815739位置的多态性,控制了α-肌动蛋白-3的合成,这是一种z结构蛋白,主要存在于快肌肉纤维中,通过加速糖酵解和促进“爆发性”强收缩为肌肉提供能量。actn3突变纯合子因为基因突变造成了α-肌动蛋白-3早期停止密码子完全缺失,导致蛋白表达丧失,使得肌肉代谢向更有效的有氧途径转移,从而提高身体内在耐力性能。最近对运动员和普通人群的研究进一步表明,actn3基因还影响骨骼肌对运动训练的适应性反应。检测actn3基因的snp是评价运动员耐力水平和训练的有效手段之一。
在snp分析中,往往根据实际样品的性质和不同实验目的来确定检测方法。snp检测方法有多种。在运动科学实验研究中,大多数检测都采用常规方法,如pcr-rflp和sanger测序法,这些方法灵敏度高,但操作复杂,分析成本较高。目前,snp检测主要用pcr、rca等dna扩增方法对样品进行信号放大,然后用实时荧光定量pcr进行信号检测,结合了荧光染料放大能力和扩增的高灵敏度,但也存在一些缺点,如操作复杂、选择性低及需要高精确度的温度循环仪器等。环介导等温扩增技术(lamp)是目前核酸分析的最灵敏的技术,最低可以检测到低于10个拷贝的dna分子,而且是等温扩增,不需要热循环过程,但存在一些不足之处,一是对单碱基错配的识别能力不高,二是需要将目标序列分为六个区域,设计四个特异性扩增引物,引物设计还要考察二级结构,保证扩增的特异性。
技术实现要素:
有鉴于此,本发明提供一种引物探针组合、试剂盒及其用于检测actn3基因突变的应用。本发明基于连接酶的lamp方法为snp检测提供了一种全新的恒温、均相、无标记、高灵敏检测技术,从而实现运动员基因型的高效检测和运动训练学评价。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了引物探针组合,所述引物包括fip、bip、primerf和primerr;所述探针包括probe-wild-a、probe-mutant-a、probe-wild-b和probe-mutant-b;
所述fip的核苷酸序列如seqidno.7所示;
所述bip的核苷酸序列如seqidno.8所示;
所述primerf的核苷酸序列如seqidno.9所示;
所述primerr的核苷酸序列如seqidno.10所示;
所述probe-wild-a的核苷酸序列如seqidno.3所示;
所述probe-mutant-a的核苷酸序列如seqidno.4所示;
所述probe-wild-b的核苷酸序列如seqidno.5所示;
所述probe-mutant-b的核苷酸序列如seqidno.6所示。
在上述研究的基础上,本发明还提供了所述的引物探针组合在制备检测actn3基因突变的试剂或试剂盒的应用。
在本发明的一些具体实施方案中,所述actn3基因突变位于rs1815739。
在上述研究的基础上,本发明还提供了所述的引物探针组合在制备检测运动员的耐力性能和/或训练成果的试剂或试剂盒中的应用。
本发明还提供了包括如权利要求1所述的引物探针组合的试剂盒。
在本发明的一些具体实施方案中,还包括ampligase热稳定dna连接酶。
本发明还提供了检测运动员的耐力性能和/或训练成果的方法,包括以下步骤:
步骤1、提取待测样品的dna;
步骤2、以所述待测样品的dna为模板,利用所述的引物探针组合或所述的试剂盒,扩增;
步骤3、根据步骤2中所述扩增的结果判定所述待测样品的耐力性能和/或训练成果。
在本发明的一些具体实施方案中,所述判定的标准为:
(1)当wild-dna探针先出现荧光信号mutant-dna探针不出现荧光信号时,样品测序结果显示snp位点存在单一的胞嘧啶c碱基信号,鉴定为actn3野生纯合子dna;
(2)当mutant-dna探针先出现荧光信号wild-dna探针不出现荧光信号,样品测序结果显示snp位点存在单一的胸腺嘧啶t碱基信号,鉴定为actn3突变纯合子;
(3)用mutant-dna探针和wild-dna探针dna检测荧光曲线poi值趋向一致时,测序结果显示snp位点同时产生胞嘧啶和胸腺嘧啶c/t碱基信号,鉴定为actn3杂合子。
在本发明的一些具体实施方案中,所述扩增包括连接反应和lamp反应;
10μl的所述连接反应的体系中包含1μl10×ampligase反应缓冲液(200mmtris-hcl,ph=8.3,250mmkcl,100mmmgcl2,5mmnad,0.1%tritonx-100),1uampligase热稳定dnaligase,1μl1μm如权利要求1所述的引物探针组合,不同浓度的1μldna或人基因组dna,5.8μlh2o,在95℃下加热3分钟,在65℃下加热30min;
所述lamp反应为:取所述连接反应的产物2μl,加入0.4μmfip和bip引物,2.5mmdntps0.8μl,2μl甜菜碱,0.8μl10×thermopol反应缓冲液(包含200mmtris-hcl,100mmkcl,100mm(nh4)2so4,20mmmgso4和1%tritonx-100、ph8.8),然后迅速加入含有0.2μl10×thermopol反应缓冲液、0.2μl20×sybrgreeni,2ubstdna聚合酶大片段和h2o的混合溶液,溶液最终体积为10μl,将最终的混合溶液在65℃下进行lamp反应,每隔1min检测和采集实时荧光强度数据。
本发明利用连接酶的连接反应,建立了一种基于连接酶的lamp检测actn3基因snp的方法。它仅需要等温条件进行扩增,同时利用ampligase热稳定dna连接酶对目标序列snp位点进行特异性识别后连接,利用连接探针的引发lamp扩增,使用荧光染料嵌入法进行实时荧光定量,避免了对探针的荧光标记,检测效率和特异性得到了大大提高,从而可以建立快速、灵敏的snp检测分析方法。我们提取了38名中国马拉松运动员全血基因组dna样品,并测定了actn3snp类型,进行了运动员snp类型和耐力指标相关性分析。该方法简化了实验步骤,可最低检测出100amdna,并成功应用于人基因组dna样品检测,可以从野生型dna中精确测量0.1.%的突变dna频率。本发明研究的基于连接酶的lamp方法为snp检测提供了一种全新的恒温、均相、无标记、高灵敏检测技术,从而实现运动员基因型的高效检测和运动训练学评价。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
图1示基于连接酶的环介导等温扩增检测actn3基因snp原理;
图2示基于连接酶的环介导等温扩增检测actn3基因snp方法的灵敏度和动态范围考察;其中,图2(a)不同浓度wild-dna产生的实时荧光曲线,wild-dna浓度分别为10pm,1pm,100fm,10fm,1fm,100am和空白样品,图2(b)实时荧光曲线的poi值和wild-dna浓度的对数(lg)的线性关系,从三次重复反应的标准偏差计算errorbars,反应在10μl溶液中进行,实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图3示基于连接酶的环介导等温扩增检测actn3基因snp方法的特异性考察;其中,图3(a)用wild型探针分别检测空白、10fmwild-dna和mutant-dna的实时荧光曲线;图3(b)用mutant型探针分别检测空白、10fmmutant-dna和10wild-dna的实时荧光曲线;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图4示混合dna样品中不同比例wild-dna的实时荧光曲线;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;其中,图4(a)示基于连接酶的环介导等温扩增检测actn3基因snp方法在过量dna(1pm)样本下分析检测snp基因频率的能力;图4(b)示基于连接酶的环介导等温扩增检测actn3基因snp方法在过量dna(10fm)样本下分析检测snp基因频率的能力;
图5示基于连接酶的环介导等温扩增检测actn3基因snp方法对人类基因组dna中snp进行测定实时荧光曲线;其中,图5(a)不同浓度基因组dna产生实时荧光曲线;基因组dna浓度从左到右依次为50ng/μl、10ng/μl、5ng/μl、2ng/μl、0.2ng/μl和空白;图5(b)实时荧光曲线中的poi值与基因组dna浓度的对数(lg)之间的线性关系;用三次重复测量的标准偏差估计误差;
图6示分别采用基于连接酶的lamp法和测序法获得的基因组dna样本检测的基因型结果;
图7示分子信标法原理示意图;
图8示taqman探针法检测snp原理示意图;
图9示荧光共轭聚合物法检测snp原理示意图;
图10示嵌入型荧光染料法结合brca检测snp原理示意图;
图11示焦磷酸测序法原理示意图;
图12示不同浓度fip和bip引物对lamp反应的实时荧光曲线;其中,(a)0.2μm;(b)0.4μm;(c)0.8μm;(d)1.6μm;实验条件:[sg]=0.4ngμl-1,[dntps]=0.2mm;
图13示不同温度对lamp反应的实时荧光曲线;其中,(a)55℃;(b)60℃;(c)65℃;(d)70℃;实验条件:[fip]=[bip]=0.4μm,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图14示不同bstdna大片段聚合酶用量对lamp反应的影响;其中,(a)1u,(b)2u,(c)4u,(d)8u;实验条件:[fip]=[bip]=0.4μm,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图15示不同浓度的探针对lamp反应的实时荧光曲线;其中,(a)4pm,(b)40pm,(c)0.4nm,and(d)4nm;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图16示不同浓度的探针对wild-dna、mutant-dna与空白样品的实时荧光曲线;其中,(a)4pm,(b)40pm,(c)0.4nm;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图17示不同连接温度对lamp反应的实时荧光曲线;其中,(a)55℃,(b)65℃,and(c)℃;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图18示不同连接温度对野生dna、突变dna目标链与空白样品的实时荧光曲线;其中,(a)55℃,(b)65℃;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm;
图19示不同dna连接酶对lamp反应的实时荧光曲线;其中,(a).0.2u,(b)0.5u,(c)1u,(d)1.5u;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm。
具体实施方式
本发明公开了一种引物探针组合、试剂盒及其用于检测actn3基因突变的应用,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
在基于连接酶的lamp方法检测actn3基因snp的试验中,设计了探针a和探针b两个茎环结构的探针(probe-wild-a、probe-wild-b),茎部单链部分与目标dna序列互补,在probe-wild-a5'端修饰了磷酸基团,设计探针互补序列时actn3基因snp位点上下游序列可以通过美国国立生物信息中心(thenationalcenterforbiotechnologyinformation,ncbi)数据库中的est数据库查询。在探针设计时尽量使两条探针与dna互补的序列tm值接近,以保证两条探针与dna杂交都处于最佳状态,采用在线核酸序列分析插件oligoanalyzer3.1(http://www.idtdna.com/calc/analyzer)对探针结构及tm值进行辅助分析;此外,两个引物fip和bip设计成分别与probe-wild-a、probe-wild-b的环部分杂交。在实验进行过程中,目标dna存在时,probe-wild-a、probe-wild-b可与变性后目标dna单链杂交。在ampligasedna连接酶的催化作用下,将探针probe-wild-b的3'端羟基和probe-wild-a的5'端磷酸基团通过磷酸二酯键连接起来,形成含有双茎环单链dna结构(doublestem-loopstructure),该结构是环介导等温扩增技术(lamp)循环扩增的起始模板。然后以双茎环单链dna结构为模板,fip和bip为引物,引发lamp反应,发生类似于传统lamp扩增中的循环扩增过程。当目标序列的snp位点和probe-wild-b不匹配时,无法产生dna连接产物,从而无法引发lamp扩增。lamp扩增时,首先双茎环单链dna结构从3'端开始自身延伸,形成发夹状dna结构(hairpindnastructure),随后引物bip与环状序列的f2部分杂交,并延伸开始链置换反应,导致发夹dna中b1和b1c部分序列的解离,解离的b1和b1c可以杂交形成茎环结构。然后,fip引物与环状序列b2c杂交,延伸合成新链,并与bip引物结合启动新一轮扩增,并且产物dna长度增加一倍,新的产物分别与茎环状结构结合启动链置换合成以这种方式重复,最后形成lamp扩增反应,扩增的最后产物是具有不同个数的茎环结构、不同长度dna的混合物,从而实现dna指数扩增。使用poi值(实时荧光曲线中的最大斜率对应的时间)定量检测目标dna链,当碱基突变位点与探针互补时,由于扩增效率高,产生指数扩增时间短,从而观察到低的poi值,同时dna和空白的poi值相差很大;当突变位点与探针不互补时,观察到高的poi值,dna和空白的poi值相差很小或趋向一致,通过样品实时荧光定量曲线poi值差异从而实现snp检测。
本发明利用连接酶反应提高了了单碱基错配的识别能力,利用lamp获得高灵敏度,从而建立了基于连接酶的lamp检测snp的新方法,该方法可以检测到浓度为100am的dna。同时我们将此方法应用于人全血基因组dna样品检测,获得了与dna测序一致的结果;该方法对基因突变具有很高的选择性,可测定0.1%突变频率。相比而言,采用taqman探针、分子信标探针等检测snp,探针合成成本较高,检测灵敏度最低为1-10fmol,所以基于连接酶的lamp方法有广泛的应用前景。例如选拔优秀运动员时,需进行能力评定,可以通过检测其血液中有氧能力、代谢效率、训练敏感性、心脏猝死和运动损伤风险等相关基因的snp类型,对其未来运动能力水平和运动风险进行预测。
本发明提供的引物探针组合、试剂盒及其用于检测actn3基因突变的应用中所用原料及试剂均可由市场购得。
2720热循环器(appliedbiosystems,美国)用于控制反应温度;stepone实时定量pcr系统(appliedbiosystems,美国)用来监测lamp反应产生的荧光。tu1901型紫外分光光度计(北京普析通用仪器有限责任公司,中国)用来分析基因组dna浓度。
bstdna聚合酶大片段(8000u/ml)购自newenglandbiolabs公司(美国)。热稳定ampligasednaligase(5000u/ml)购自epicentretechnologies(美国)。甜菜碱购自sigma公司(美国)。sybrgreen(20×dmso缓冲液配制,20μg/ml)购自bio-visionbiotechnology(中国)。基因组dna提取试剂盒购自天根生物公司(中国)。合成的dna、碱性磷酸酶(5000u/ml)、dntps购自宝生物科技有限公司(大连,中国),实验所用的dna序列见表1。下划线标出的碱基为突变点。突变型和野生型dna为人类actn3基因片段。所有溶液均用去离子灭菌水配制。其它试剂均为分析纯。
表1野生型和突变型dna、各种探针、引物的序列
本发明利用连接酶反应提高了单碱基错配的识别能力,利用lamp获得高灵敏度,从而建立了基于连接酶的lamp检测snp的新方法,该方法可以检测到浓度为100am的dna。同时我们将此方法应用于人全血基因组dna样品检测,获得了与dna测序一致的结果;该方法对基因突变具有很高的选择性,可测定0.1%突变频率。相比而言,采用taqman探针、分子信标探针等检测snp,探针合成成本较高,检测灵敏度最低为1-10fmol,所以基于连接酶的lamp方法有广泛的应用前景。例如选拔优秀运动员时,需进行能力评定,可以通过检测其血液中有氧能力、代谢效率、训练敏感性、心脏猝死和运动损伤风险等相关基因的snp类型,对其未来运动能力水平和运动风险进行预测。
下面结合实施例,进一步阐述本发明:
实施例1
(1)基因组dna的提取
人基因组dna样品从38名中国马拉松运动员血液中提取获得,采用天根生物公司(中国)基因组dna提取试剂盒提取,提取实验操作步骤按试剂盒说明书,并用tu1901型紫外分光光度计在260nm波长下分析样品浓度。
(2)连接反应
10μl的反应体系中包含1μl10×ampligase反应缓冲液(200mmtris-hcl,ph=8.3,250mmkcl,100mmmgcl2,5mmnad,0.1%tritonx-100),1uampligase热稳定dnaligase,1μl1μmdna探针(dna碱基序列见表1),不同浓度的1μldna或人基因组dna,5.8μlh2o,将混合物溶液放入2720热循环器中,在95℃下加热3分钟,在65℃下加热30分钟。
(3)lamp反应
取以上连接产物2μl,加入0.4μmfip和bip引物,2.5mmdntps0.8μl,2μl甜菜碱,0.8μl10×thermopol反应缓冲液(包含200mmtris-hcl,100mmkcl,100mm(nh4)2so4,20mmmgso4和1%tritonx-100、ph8.8),然后迅速加入含有0.2μl10×thermopol反应缓冲液、0.2μl20×sybrgreeni,2ubstdna聚合酶大片段和h2o的混合溶液。溶液最终体积为10μl,将最终的混合溶液立即放入stepone实时荧光定量pcr仪中,在65℃下进行lamp反应,每隔1分钟检测和采集实时荧光强度数据。
(4)测序分析
为了获得人基因组dna的测序结果,首先制备dna测序的模板,包含5μl10×extaq缓冲液、5μl2.5mmdntps、1μl10μm的引物f和引物r、1μltakaraextaqhs聚合酶、1μl基因组dna和h2o,总体积50μl,放入2720pcr系统进行pcr扩增。在98℃反应10s,55℃反应30s,72℃反应1分钟,反应循环30次。为了验证扩增产物的正确性,取1μl的反应产物进行3%琼脂糖凝胶电泳。用碱性磷酸酶和核酸外切酶i纯化pcr产物,并在rs1815739snp位点进行测序,测序工作送至宝生物科技有限公司完成。
实施例2
在最佳的实验条件下(40pm连接探针浓度、1uampligasedna连接酶、0.4μmfip、bip引物浓度、2ubstdna聚合酶和65℃的连接温度),按上述实验过程对不同浓度的wild-dna样品进行基于连接酶的lamp反应,并记录其实时荧光强度随反应时间变化的曲线。
由图2所示,图2(a)示不同浓度wild-dna产生的实时荧光曲线,wild-dna浓度分别为10pm,1pm,100fm,10fm,1fm,100am和空白样品,图2(b)示实时荧光曲线的poi值和wild-dna浓度的对数(lg)的线性关系,从三次重复反应的标准偏差计算errorbars,反应在10μl溶液中进行,实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm。
通过基于连接酶的lamp反应和实时荧光强度测量,wild-dna在100am-10pm浓度范围内可以被定量测定,最低可检测100amwild-dna样品(相当于在10μl体积溶液中的600个拷贝dna分子)。用poi(实时荧光曲线上最大斜率对应的时间点)定量检测目标dna,poi值在从100am-10pm的范围内和目标野生dna浓度的对数(lg)成良好的线性关系。相关系数方程为poi=51.9+7.81lgc(m),相关系数r=0.9996,线性范围在6个数量级以上,表明了此方法具有高灵敏度和较宽的动态范围。
实施例3
为了评价分析方法的特异性,我们选择同浓度(10fm)具有高序列同源性的wild-dna和mutantdna作为模型。在同等实验条件下,进行基于连接酶的lamp反应,并分别测量实时荧光强度曲线。
图3所示,图3(a)用wild型探针分别检测空白、10fmwild-dna和mutant-dna的实时荧光曲线;图3(b)用mutant型探针分别检测空白、10fmmutant-dna和10wild-dna的实时荧光曲线;实验条件:[fip]=[bip]=0.4μm,[bstdnapolymerase]=0.2uμl-1,[sg]=0.4ngμl-1,[dntps]=0.2mm。
当用wild探针测定样品时,在wild-dna荧光强度曲线对应的poi值mutant-dna不产生可测的荧光强度。同理,在用mutant探针测定样品时,mutant-dna荧光强度曲线对应的poi值,没有观察到wild-dna和空白的荧光信号,wild-dna和mutantdna有一个碱基的序列差别。因此该方法具有较高的特异性,可以明确区分dna序列中一个碱基的差异,从而非常有效地检测单碱基突变。
实施例4
snp的定量检测数据可应用于snp等位基因频率的估计,这对于评价acnt3基因型与运动员耐力表现之间的关系具有重要意义。为此利用基于连接酶lamp方法的特异性和灵敏度的优点,我们在过量dna样本下分析该方法检测snp基因频率的能力。
将wild-dna和mutant-dna以不同比例混合(0、0.1、1、10和100%)作为dna样本。样品中dna的总浓度控制为1pm和10fm。然后对样品经过连接、lamp反应和实时荧光定量检测,获得荧光强度与突变频率之间的关系。混合样品检测结果如图4a、图4b所示,突变频率在1%-100%范围内与poi值成良好线性关系。当总浓度固定在1pm(图4a)时,在1000倍高浓度的突变dna存在下,可以检测到低至0.1%的野生dna。当总浓度为10fm时,可以检测到低至1%的野生dna。结果表明,该方法可成功地应用于dna样品中snp基因频率的测定。结果显示,该方法适合对微量dna混合样品中的snp定量检测。
实施例5
为了进一步验证方法的实用性,按上述实验过程对人类基因组dna中snp进行测定,如图5所示。图5(a)不同浓度基因组dna产生实时荧光曲线;基因组dna浓度从左到右依次为50ng/μl、10ng/μl、5ng/μl、2ng/μl、0.2ng/μl和空白;图5(b)实时荧光曲线中的poi值与基因组dna浓度的对数(lg)之间的线性关系。本方法检测0.2ng/μl-50ng/μl浓度范围内的基因组dna的实时荧光曲线具有良好的线性关系,可以用于定量检测浓度低至0.2ng/μl的人基因组dna样品,线性方程poi=-21.1+8.82lgc(m),相关系数r=0.929。根据dsdna摩尔浓度和质量换算公式0.2ng/μl=3.04fmoldsdna。用连接酶lamp方法检测实际样品时灵敏度低于合成dna样品,推测由于基因组dna样品比合成dna样品成分复杂,且含有更多数量级的dna序列所致。
实施例6
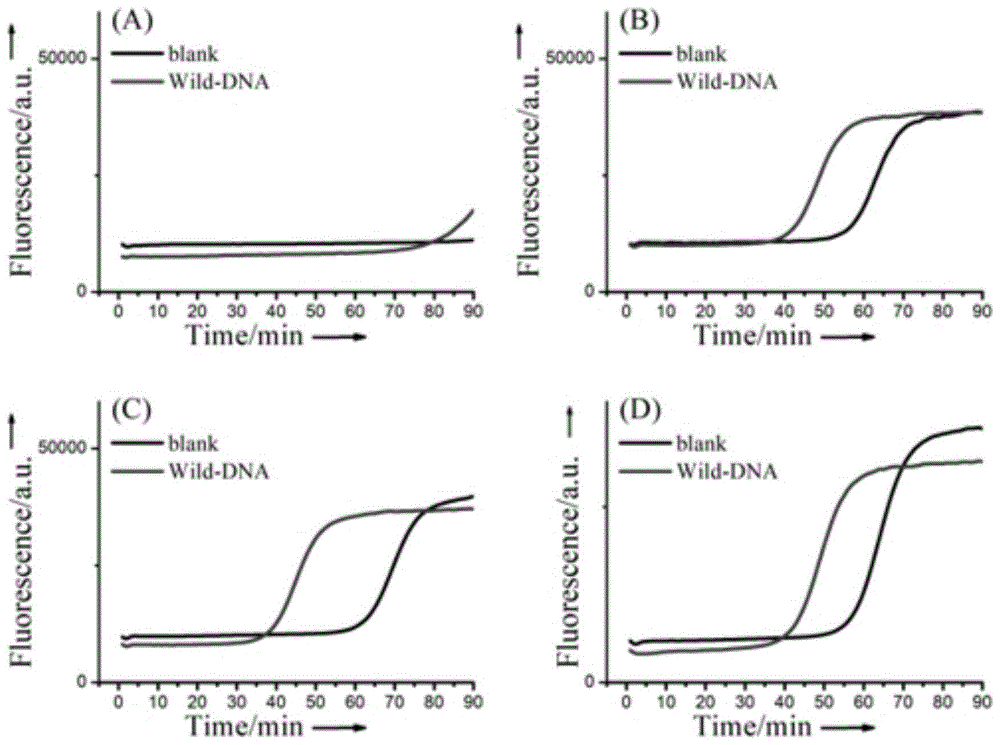
为了进一步验证方法的可靠性,分别使用wild-dna探针和mutant-dna探针,采用基于连接酶的lamp法和sanger测序测定同一人基因组dna样品,基因组dna浓度为20ng/μl,进行结果比对。如图6所示。
当wild-dna探针先出现荧光信号mutant-dna探针不出现荧光信号时,样品测序结果显示snp位点存在单一的胞嘧啶(c)碱基信号,鉴定为actn3野生纯合子dna;当mutant-dna探针先出现荧光信号wild-dna探针不出现荧光信号,样品测序结果显示snp位点存在单一的胸腺嘧啶(t)碱基信号,鉴定为actn3突变纯合子;用mutant-dna探针和wild-dna探针dna检测荧光曲线poi值趋向一致时,测序结果显示snp位点同时产生胞嘧啶和胸腺嘧啶(c/t)碱基信号,鉴定为actn3杂合子(图6c)。实验结果表明,通过基于连接酶的lamp法和sanger测序测定基因组dna样本snp结果一致,证明本文所开发的检测方法可能成为基因研究的一种有前途的技术,可用于已知序列的基因分型。
对比例1lamp反应fip和bip引物浓度不同对检测灵敏度的影响
本发明采用了以10fmwild-dna为样品,以wild探针作为反应底物,进行lamp反应参数的优化实验。图12显示了lamp反应中fip和bip引物浓度对检测的影响。当fip、bip引物浓度增加时,反应加速,但空白和wild-dna的实时荧光曲线poi差值降低;当引物浓度大于0.4μm时,随着引物浓度增加,空白和wild-dna实时荧光曲线poi差值减小,在fip和bip引物的浓度为0.4μm时,空白和wild-dna的poi差值最大,因此,fip和bip引物的最适反应浓度为0.4μm。
对比例2lamp反应不同温度对检测灵敏度和特异性的影响
lamp反应温度对反应的特异性和效率产生影响,反应温度过低,容易产生fip、bip引物的非特异性杂交,从而引起线性扩增;反应温度过高,bstdna聚合酶大片段的活性受到限制,影响指数扩增的效率。为了探讨lamp反应温度对检测过程的影响,分别在55℃、60℃、65℃、70℃进行lamp扩增实验。结果如图13所示,当反应温度为55℃时,反应在40分钟内wild-dna产生明显的非特异线性扩增;随着lamp反应温度从55℃提高到65℃,wild-dna与空白的poi值差异逐渐增大,非特异扩增减少。当温度达到70℃时,由于温度过高,wild-dna和空白均不发生指数扩增。综合考虑poi差值和非特异性扩增影响,选择65℃作为lamp反应的最适温度。
对比例3
lamp反应bstdna大片段聚合酶用量的影响
用来催化lamp链顶替和置换反应的bstdna聚合酶用量是影响检测效果的一个关键因素。过量的bstdna聚合酶大片段的存在会引起引物的非特异性杂交和扩增,从而造成假阳性;过少的bstdna聚合酶大片段使得连接产物剩余,使得lamp反应扩增效率变低,指数扩增时间延长。图14显示了lamp反应中bstdna大片段聚合酶用量对检测的影响。bstdna大片段聚合酶的用量从2u增加到8u的过程中,wild-dna的poi值降低,但与空白的poi值差异减小,可能是bstdna聚合酶大片段过量,在增加扩增效率的同时,引起了空白样品中引物的非特异性杂交和扩增。当bstdna聚合酶大片段用量从2u降低至1u时,wild-dna和空白的poi差值减小,当bstdna聚合酶大片段用量为2u时,wild-dna和空白的poi差值最大。因此,选用2ubstdna聚合酶大片段进行检测最为合适。
连接反应中探针浓度对反应灵敏度和特异性的影响
在连接反应中,采用不同浓度的探针检测wild-dna,探针的浓度影响连接反应的特异性。探针过量会产生非特异性扩增,从而降低检测的特异性;探针浓度过低会使得扩增效率降低,指数扩增时间增加。如图15所示,当探针浓度下降到4pm时,因为探针不够形成足够的连接产物使得lamp指数扩增时间增加。当探针浓度从40pm升高至4nm时,随着探针浓度的升高指数扩增时间缩短;此外,在探针浓度40pm至0.4nm之间,wild-dna与空白的poi值差异增大;当探针浓度从0.4nm升高到4nm时,由于探针过量使得wild-dna指数扩增的时间缩短,但和空白的poi差异降低。
此外,反应中过量的连接探针会导致单碱基错配,非特异性连接几率增加,从而影响对wild-dna和mutant-dna检测特异性。如图16所示,当探针浓度从4pm增加至40pm时,因为探针浓度增加,lamp扩增效率增加,wild-dna和mutant-dna及空白的poi差值增加,snp检测的特异性升高;当探针浓度从4pm增加至40pm时,由于探针过量导致非特异性连接几率增加,wild-dna和mutant-dna的poi差值降低,snp检测特异性下降。综合考虑上述因素,选用40pm探针浓度用于检测。
对比例4连接反应中温度对反应灵敏度的影响
连接反应温度和lamp法的灵敏度及选择性密切相关。一般来说,较低的连接温度可以提高连接反应的敏感性,但反应特异性较差;而较高的连接温度会明显降低非匹配探针和目标序列的的杂交效率,从而提高连接反应的特异性,但却降低扩增效率,如果温度再提高,就可能会超过探针杂交的tm值而影响到匹配探针和目标序列的杂交效率。为了研究连接反应温度对检测过程的影响,在连接温度分别为55℃、65℃和70℃时检测10fmwild-dna样品。如图17所示,当连接温度从55℃升高到65℃时,wild-dna与空白之间的poi值差异减小,当连接温度为70℃时,探针和目标序列杂交因为反应温度过高受到抑制不产生指数扩增。如图18所示,当连接温度为55℃时,wild-dna和mutantdna的poi值几乎一致,可能由于过低的连接温度降低了反应的特异性。当连接温度为65℃时,wild-dna和mutantdna的poi差异增大。综上所述,选择65℃作为连接反应最适温度用于检测。
对比例5连接反应中ampligasedna连接酶用量的对反应灵敏度的影响
ampligasedna连接酶用量影响连接反应效率和特异性。图19显示ampligasedna连接酶用量对检测的影响。当连接酶用量为0.2u时,由于参与反应的连接酶量不足,反应时间增加,wild-dna和空白样品在90分钟内均未检测到指数扩增,不利于对目标序列检测。当连接酶用量从0.5u增加至1u时,随着连接酶用量的增加,连接效率提高,wild-dna的poi值增加,和空白poi差值也同时增大,wild-dna目标链与空白样品之间的poi值差异在1u时达到最大。当连接酶用量1u增加至1.5u时,检测所需的反应时间并未因连接酶用量增加而增加,因为此时ampligasedna连接酶已经过量,反而由于ampligasedna连接酶促进非特异性连接而使得wild-dna和空白的poi值差异减小,综上所述,选择1uampligasedna连接酶用于检测。反应参数优化结果统计见表2。
表2反应参数优化结果
对比例6
根据lcr反应设计了两对探针。每对探针包含和目标序列上游和下游互补的两个探针,其中一个探针在3’端有磷酸修饰,另一个探针在5’端标记有荧光素。lcr反应时,当探针和目标序列互补两个相邻探针连接成一条dna链,在dna链5’端带有荧光素标记,3’端具有抗核酸外切酶降解的磷酸修饰,然后连接产物进行lcr扩增,产生大量的相同产物,加入pfp后,由于pfp与dna之间的强静电相互作用,可以观察到从pfp到荧光标记dna的有效荧光共振能量转移(fret)。如果目标序列发生snp位点突变,使得dna连接酶无法形成探针的连接产物,加入核酸外切酶i和iii使探针从5’-3’端酶切降解,导致加入pfp后不能产生fret信号,从而成功地实现了lcr均相检测snp。该方法灵敏度高、特异性强,可检测1fmol(600zmol)dna分子。与本发明比较而言,高出了一个数量级。
表3野生型和突变型dna、挂锁探针和各种引物的序列
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>申请人
<120>引物探针组合、试剂盒及其用于检测actn3基因突变的应用
<160>10
<170>siposequencelisting1.0
<210>1
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>1
caaggcaacactgcccgaggctgaccgagagcgaggtgccatcatgggcat51
<210>2
<211>51
<212>dna
<213>人工序列(artificialsequence)
<400>2
caaggcaacactgcccgaggctgactgagagcgaggtgccatcatgggcat51
<210>3
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>3
gtcagcctcgggcagtgttgttttatcgtcgtgactgtttgtaata46
<210>4
<211>32
<212>dna
<213>人工序列(artificialsequence)
<400>4
ggacagagccccgcactttcagtcacgacgat32
<210>5
<211>78
<212>dna
<213>人工序列(artificialsequence)
<400>5
cgacagcagaggatttgttgtgtggaagtgtgagcggattttcctctgctgtcgttttat60
gatggcacctcgctctcg78
<210>6
<211>78
<212>dna
<213>人工序列(artificialsequence)
<400>6
cgacagcagaggatttgttgtgtggaagtgtgagcggattttcctctgctgtcgttttat60
gatggcacctcgctctca78
<210>7
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>7
atcgtcgtgactgaaagtgcggggctctgtcctattac38
<210>8
<211>38
<212>dna
<213>人工序列(artificialsequence)
<400>8
cgacagcagaggatttgttgtgtggaagtgtgagcgga38
<210>9
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>9
ctgttgcctgtggtaagtggg21
<210>10
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>10
tggtcacagtatgcaggaggg21
- 还没有人留言评论。精彩留言会获得点赞!
- 一种用于评估皮肤抗过敏情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤防御情况的引物和探针及试剂盒的制作方法
- 一种荧光定量PCR检测猪delta冠状病毒的引物、探针、试剂盒及方法
- 一种快速区分HP-PRRS疫苗GDr180株与野毒株的引物、探针及方法
- 一种用于检测样品中猪凸隆病毒的荧光rt-pcr引物和探针和试剂盒及检测方法
- 一种阿胶中驴、马、牛和猪源性巢式荧光pcr检测引物、探针、试剂盒、检测方法及应用
- 一种用于评估皮肤防晒情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤抗皱情况的引物和探针及试剂盒的制作方法
- 一种用于检测car-t转导效率的引物、探针和方法
- 用于微嵌合检测和个体识别的引物、探针、试剂盒及方法