一种多拉司琼N-氧化物及其制备方法和用途与流程
一种多拉司琼n-氧化物及其制备方法和用途
技术领域
1.本发明涉及一种化合物及其制备方法和用途,具体涉及一种多拉司琼n-氧化物及其制备方法和用途,属药物化学领域。
背景技术:
2.恶心、呕吐是肿瘤化疗中最常见的不良反应,75%以上的化疗病人均会出现不同程度的恶心、呕吐。严重的呕吐不仅使患者感到不适、意志消沉,而且会导致电解质失衡、营养缺乏,严重削弱机体自身抵抗力,甚至部分病人常因呕吐反应对化疗产生恐惧,以致中断治疗,贻误控制肿瘤的时机,最终威胁到生命。止吐药不但能改善患者的生活质量,同时也能确保化疗的顺利进行。
3.甲磺酸多拉司琼是一种止吐药,属于新型高选择性、高亲和性5-ht3受体拮抗剂,它通过竞争5-ht,有效地阻滞外周和中枢的5-ht3受体,从而抑制或减少恶心和呕吐反射的发生,具有高效、耐受性好、无锥体外系副作用等优点,在临床应用中具有广阔的市场前景。甲磺酸多拉司琼是多拉司琼的甲磺酸盐,多拉司琼的结构式如下:
[0004][0005]
但是,多拉司琼在光照下容易发生氧化,有关物质有一定程度的增加,至今为止,未见有关多拉司琼n-氧化产物的分离、结构确认及其用途研究的相关报道。
技术实现要素:
[0006]
本发明的目的在于提供一种多拉司琼n-氧化物,具有式(ⅰ)所示结构:
[0007][0008]
其化学名为内-六氢-8-(3-吲哚甲酰氧基)-2,6-二亚甲基-2h-喹嗪-3(4h)酮-5-氮氧化物。
[0009]
本发明的另一目的在于提供一种制备多拉司琼n-氧化物的方法,由多拉司琼或其盐在反应溶剂中与氧化剂反应,然后加还原剂淬灭反应得到式(ⅰ)所示多拉司琼n-氧化物。
[0010]
进一步地,所述反应溶剂选自甲醇、乙醇、四氢呋喃、二氯甲烷中的一种或多种的混合;所述氧化剂选自双氧水、间氯过氧苯甲酸、过氧化氢异丙苯中的一种或多种的混合;所述还原剂选自亚硫酸钠或亚硫酸氢钠或亚硫酸钠和亚硫酸氢钠的混合物。
[0011]
进一步地,所述多拉司琼与所述氧化剂的摩尔投料比为1:1.0~20,所述还原剂与所述氧化剂的摩尔投料比为1:0.5~2。
[0012]
更进一步地,所述多拉司琼与所述氧化剂的摩尔投料比为1:2~10;所述还原剂与所述氧化剂的摩尔投料比为1:1。
[0013]
进一步地,所述与氧化剂反应的反应温度控制在-10℃~70℃,所述加入还原剂淬灭反应的反应温度控制在-10℃~70℃。
[0014]
更进一步地,所述与氧化剂反应的反应温度控制在0℃~50℃,所述加入还原剂淬灭反应的反应温度控制在0℃~50℃。
[0015]
在一实施方案中,所述的制备方法还包括使用高效制备液相色谱梯度洗脱进行分离纯化的步骤,其色谱条件为:采用碳十八键合硅胶色谱柱;流动相a为0.03%氨水溶液,流动相b为乙腈;检测波长为210
±
10nm。
[0016]
本发明的多拉司琼n-氧化物,可用作对照品用于控制多拉司琼游离碱或其药学上可接受盐及其制剂的质量,并用于检测多拉司琼游离碱或其药学上可接受盐及其制剂的有效期。因此,本发明的另一目的在于提供多拉司琼n-氧化物用于制备多拉司琼药物或其制剂的对照品中的应用。
[0017]
本发明的又一目的是提供一种药物组合物,所述药物组合物含有多拉司琼或其药学上可接受的盐和质量百分含量不高于0.4%的式(ⅰ)所示结构化合物,即本发明多拉司琼n-氧化物:
[0018][0019]
所述多拉司琼药学上可接受的盐选自马来酸盐、甲磺酸盐、磺酸盐、硫酸盐、盐酸盐、氢溴酸盐、磷酸盐、硝酸盐、对甲苯磺酸盐、酒石酸盐、富马酸盐、乙酸盐、甲酸盐、苯甲酸盐、肉桂酸盐、丁二酸盐、丙二酸盐、枸橼酸盐中的任一种或其组合。
[0020]
进一步地,所述药物组合物中多拉司琼或其药学上可接受盐的含量不低于90%,优选不低于95%,更优选不低于98%。
[0021]
进一步地,式(ⅰ)所示结构化合物的含量不高于0.3%,优选不高于0.2%,更优选不高于0.1%。
[0022]
进一步地,多拉司琼或其药学上可接受的盐与式(ⅰ)化合物之间的重量比不低于300:1,优选不低于400:1,更优选不低于600:1。
[0023]
在一实施方案中,所述药用组合物含有含量不低于98%的多拉司琼或其药学上可接受的盐、含量不高于0.10%的式(ⅰ)所示多拉司琼n-氧化物、含量不高于1.0%的多拉司琼有关物质a,余量为其他有关物质。
[0024]
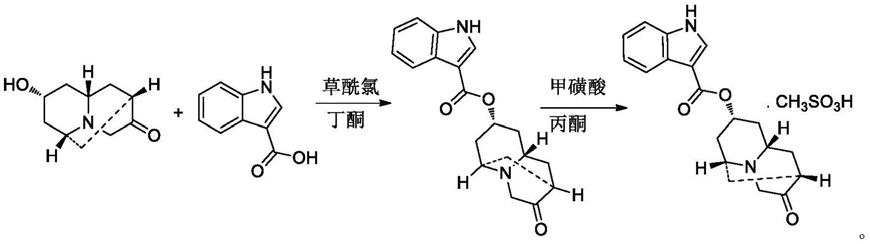
上述药物组合物的制备方法为:在避光及氮气或惰性气体保护条件下,内-六氢-8-羟基-2.6-亚甲基-2h-喹嗪-3(4h)-酮或其盐与吲哚-3-甲酸反应,制备多拉司琼,多拉司琼再进一步与酸反应,制备药学上可接受的盐,如:
[0025][0026]
本发明还提供一种药物制剂,其包含上述药物组合物,以及任选的药学上可接受的载体。
[0027]
进一步地,本发明提供药物制剂,其中多拉司琼或其药学上可接受的盐与式(ⅰ)化合物之间的重量比不低于300:1,优选不低于400:1,更优选不低于600:1。
[0028]
本发明的药物制剂可为本领域熟知的各种剂型。适合于本发明的剂型选自口服制剂、外用制剂或注射剂,优选为口服制剂或注射剂,更优选为注射剂。所述注射剂选自针剂(注射液)、输液、冻干粉针或无菌分装制剂,优选为注射液。可采用本领域熟知的制剂技术手段制备得到本发明的药物制剂。
[0029]
本发明所述的药学上可接受的载体为本领域熟知的用于制备所述制剂的常用赋形剂或辅料。所述注射剂常用的赋形剂或辅料包括但不限于:抗氧剂,例如酚类如丁羟基茴香醚(bha)、丁羟基甲苯(bht)、去甲二氢愈创木酸(ndga),含硫化合物如硫二丙酸、亚硫酸盐、亚硫酸氢盐、二硫代氨基甲酸盐,亚硫酸钠、亚硫酸氢钠、焦亚硫酸钠、硫代硫酸钠;有机酸/醇/酯类如抗坏血酸、枸橼酸、苹果酸、山梨醇、甘油、丙二醇、抗坏血酸棕榈酸酯,酯类如氢醌、羟基香豆素、维生素e,胺类如乙醇胺、豆磷脂、脑磷脂、植物磷脂或动物磷脂,无机酸或其盐、磷酸或其盐、亚磷酸或其盐;渗透压调节剂,例如氯化钠、葡萄糖、氯化钾、氯化镁、氯化钙、山梨醇、甘露醇等,优选为氯化钠或葡萄糖;抑菌剂,例如0.5%苯酚、0.3%甲酚、0.5%三氯叔丁醇;ph调节剂,例如盐酸、酒石酸、柠檬酸、氢氧化钾、氢氧化钠、磷酸二氢钠、磷酸氢二钠、醋酸、醋酸钠、乳酸、枸橼酸、枸橼酸钠、碳酸氢钠、碳酸钠任一种或其组合;乳化剂,例如聚山梨酯-80、没酸山梨坦、普罗尼克f-68、卵磷酯、豆磷脂;增溶剂,例如吐温-80、胆汁、甘油、丙二醇、卵磷脂、聚氧乙烯蓖麻油等;填充剂或赋形剂,例如乳糖、甘露醇、山梨醇、右旋糖酐等。
[0030]
所述注射剂选自注射液、注射用无菌粉末与注射用浓溶液,可用于肌内注射、静脉注射、静脉滴注等。本发明注射剂的规格选自有1ml、2ml、5ml、10ml、20ml、50ml、100ml、200ml、250ml或500ml。
[0031]
本发明的药物制剂可为单位制剂,以多拉司琼计,含有0.01g~5g的本发明药物组合物作为必需的活性成分,优选为0.01~3g,更优选为0.0125g、0.04g、0.08g、0.1g、0.16g、0.2g、0.25g、0.32g、0.4g、0.5g、0.75g、1g、1.25g、1.5g、1.75g、2g或2.5g。
[0032]
特别地,本发明还提供一种甲磺酸多拉司琼注射液的制备方法,是将上述药物组合物与药学上可接受的注射液载体混合进行制备。
[0033]
进一步地,所述的药学上可接受的注射液载体选自抗氧剂、抑菌剂、ph调节剂的任一种或其组合。
[0034]
进一步地,所述的抗氧剂选自酚类如丁羟基茴香醚(bha)、丁羟基甲苯(bht)、去甲
二氢愈创木酸(ndga),含硫化合物如硫二丙酸、亚硫酸盐、亚硫酸氢盐、二硫代氨基甲酸盐,亚硫酸钠、亚硫酸氢钠、焦亚硫酸钠、硫代硫酸钠;有机酸/醇/酯类如抗坏血酸、枸橼酸、苹果酸、山梨醇、甘油、丙二醇、抗坏血酸棕榈酸酯,酯类如氢醌、羟基香豆素、维生素e,胺类如乙醇胺、豆磷脂、脑磷脂、植物磷脂或动物磷脂,无机酸或其盐、磷酸或其盐、亚磷酸或其盐。
[0035]
进一步地,所述的ph调节剂选自盐酸、酒石酸、柠檬酸、氢氧化钾、氢氧化钠、磷酸二氢钠、磷酸氢二钠、醋酸、醋酸钠、乳酸、枸橼酸、枸橼酸钠、碳酸氢钠、碳酸钠中的任一种或其组合。
[0036]
进一步地,所述的抑菌剂选自0.5%苯酚、0.3%甲酚、0.5%三氯叔丁醇中的任一种或其组合。
[0037]
本发明还提供了上述药物组合物、药物制剂在用于制备止吐药物中的应用。
[0038]
发明人在研究中惊喜地发现,本发明的多拉司琼n-氧化物具有较强的抗肿瘤活性。因此,本发明的另一目的在于提供多拉司琼n-氧化物或其组合物在用于制备抗肿瘤药物中的应用,并提供一种新型的抗肿瘤药物组合物,所述组合物中含有有效量的多拉司琼n-氧化物。
[0039]
本发明式(ⅰ)所示多拉司琼n-氧化物的结构,经核磁共振谱和质谱确证,解析如下(数据来源于本发明实施例1制备的多拉司琼n-氧化物样品的检测结果):
[0040]
1.质谱数据如下表1所示:
[0041]
表1本发明多拉司琼n-氧化物的质谱数据
[0042][0043]
质谱(ms)显示本品的[m+h]
+
峰的质荷比为341,表明本品的分子量为340,相比多拉司琼分子量(324)多16,与氧的原子量相同;质谱信号实测值与理论值偏差很小,与该多拉司琼n-氧化物分子式(c
19
h
20
n2o4)相符。
[0044]
2.核磁共振氢谱
[0045][0046]
氢谱数据,如下表2所示:
[0047]
表2本发明多拉司琼n-氧化物的核磁共振氢谱数据
[0048][0049][0050]
注:s-单峰,d-双峰,m-多重峰,t-三重峰
[0051]1h-nmr数据有合理归属,符合氢谱化学位移规律,与该多拉司琼n-氧化物的结构式相符。
[0052]
将上述核磁共振氢谱与多拉司琼游离碱核磁共振氢谱做对比分析,h-12,h-26化学位移由氧化前δ3.2719~3.2807跃迁至δ3.9497和δ4.1503;h-7化学位移由δ3.3625跃迁至δ5.8729~5.9121和δ6.0722~6.1146;h-10化学位移由δ2.5913~2.6464跃迁至δ3.6340~3.7364;h-27化学位移由δ2.4056~2.4352跃迁至δ3.3310,被包裹在溶剂峰中;h-19属于活泼氢,在氘代甲醇中没有出峰;该化学位移跃迁分析符合n-4上氮被氧化后对结构影响。
[0053]
综合上述,化合物的nmr氢谱数据有合理的归属,符合氢谱化学位移规律,与多拉司琼n-氧化物的结构式相符。
[0054]
因此,根据质谱、核磁共振谱综合分析,该多拉司琼n-氧化物即为上述式(ⅰ)所示化学结构。
[0055]
在本发明式(ⅰ)所示多拉司琼n-氧化物,通过在法国eurofins-cerep sa公司作活性研究发现,其对激酶lyn和mst1均有较强的抑制作用,表明其具有抗肿瘤作用。
[0056]
lyn是src家族激酶的成员之一,属于非受体型酪氨酸激酶,通过调节酪氨酸磷酸化状态来决定信号转导分子的结合与解离,参与信号传导作用。与多种恶性肿瘤如白血病、淋巴瘤、前列腺癌、结肠癌、乳腺癌等疾病密切相关。
[0057]
mst1是hippo信号通路的核心组件,其通过磷酸化、二聚化以及核内外定位等方式,在细胞分化、控制稳态、促进细胞黏附和迁移、抑制自噬、促进凋亡等多种生理活动中发挥重要作用。与急性白血病、湿疹血小板减少伴免疫缺陷综合症、常染色体隐性遗传的联合免疫缺陷病、前列腺癌、胃癌、乳腺癌、宫颈癌等疾病密切相关。
[0058]
本活性研究采用staurosporine作阳性对照。staurosporine是一种非选择性蛋白激酶抑制剂,为激酶实验中常用的阳性对照。
附图说明
[0059]
图1为式(ⅰ)所示结构化合物的hplc图;
[0060]
图2为式(ⅰ)所示结构化合物的1h-nmr谱图;
[0061]
图3为式(ⅰ)所示结构化合物的质谱图。
具体实施方式
[0062]
下面结合试验例和实施例对本发明作进一步说明,可使本领域专业技术人员更全面地理解本发明,但不以任何方式限制本发明。
[0063]
实施例1本发明多拉司琼n-氧化物的制备
[0064]
将多拉司琼(10g,0.0309mol)、甲醇(200ml)和30%双氧水(50ml,0.5mol)混合搅拌,加热50℃反应8小时;滴加入亚硫酸钠(63g,0.5mol)溶于水500ml的溶液。减压蒸馏除去甲醇,剩余水溶液过滤,滤饼即为粗品,样品采用制备液相(色谱柱为碳十八键合硅胶柱,流动相a:0.03%水溶液;b:乙腈,梯度洗脱,10%~40%b,60分钟;流速80ml/min)分离得到产物0.65g,hplc纯度97.7%。
[0065]
实施例2本发明多拉司琼n-氧化物的制备
[0066]
将多拉司琼(10g,0.0309mol)、乙醇(200ml)和过氧化氢异丙苯(4.72g,0.031mol)混合搅拌,搅拌,加热70℃反应8小时;滴加入亚硫酸氢钠(3.23g,0.031mol)溶于水250ml的溶液。减压蒸馏除去有机相,剩余水溶液过滤,滤饼即为粗品。样品采用制备液相(色谱柱为碳十八键合硅胶柱,流动相a:0.03%水溶液;b:乙腈,梯度洗脱,10%~40%b,60分钟;流速80ml/min)分离得到产物0.55g,hplc纯度96.5%。
[0067]
实施例3本发明多拉司琼n-氧化物的制备
[0068]
将多拉司琼(10g,0.0309mol)、四氢呋喃(200ml)混合搅拌,分批加入间氯过氧苯甲酸(8.63g,0.05mol),20℃反应12小时;滴加入亚硫酸氢钠(5.20g,0.05mol)溶于水200ml的溶液,减压蒸馏除去四氢呋喃,剩余水溶液过滤,滤饼即为粗品。样品采用制备液相(色谱柱为碳十八键合硅胶柱,流动相a:0.03%水溶液;b:乙腈,梯度洗脱,10%~40%b,60分钟;流速80ml/min)分离得到产物0.45g,hplc纯度97.1%。
[0069]
实施例4本发明多拉司琼n-氧化物的制备
[0070]
将多拉司琼(10g,0.0309mol)、二氯甲烷(200ml)混合搅拌并降温至-10℃,分批加入间氯过氧苯甲酸(17.26g,0.1mol),搅拌,保温-10℃反应48小时。滴加入亚硫酸氢钠(10.4g,0.1mol)溶于水150ml的溶液,减压蒸馏除去二氯甲烷,剩余水溶液过滤,滤饼即为粗品。样品采用制备液相(色谱柱为碳十八键合硅胶柱,流动相a:0.03%水溶液;b:乙腈,梯度洗脱,10%~40%b,60分钟;流速80ml/min)分离得到产物0.47g,hplc纯度96.4%。
[0071]
实施例5本发明多拉司琼n-氧化物的活性研究
[0072]
实验方法:
[0073]
10μm本发明多拉司琼n-氧化物与阳性staurosporine分别与0.2mg/ml lyn激酶在hepes溶液(ph7.4)中,于37℃预孵育15分钟后,加入底物(0.2mg/ml poly(glu,tyr))、10mm atp和0.25mci*γ-32p]-atp继续孵育30分钟。加入3%的磷酸溶液终止反应。检测[32p]poly(glu:tyr)形成量,并根据式1、式2分别计算激酶活性以及化合物抑制率。
[0074]
10μm本发明多拉司琼n-氧化物与阳性staurosporine分别与0.25u/ml mst1激酶
在mops溶液(ph7.2)中,于37℃预孵育15分钟后,加入50μg/ml髓磷脂碱性蛋白(mbp)、10μm atp和0.25μci*γ-32p]-atp继续孵育30分钟。加入3%的磷酸溶液终止反应。检测[32p]mbp形成量,并根据式1、式2分别计算激酶活性以及化合物抑制率。
[0075]
激酶活性%=(待测
count-阳性
count
)/(溶媒
count-阳性
count
)*100%
ꢀꢀꢀ
(式1)
[0076]
抑制率%=100-激酶活性%(式2)
[0077]
其中待测
count
为待测化合物底物同位素含量,阳性
count
为staurosporine底物同位素含量,溶媒
count
为溶媒对照底物同位素含量。
[0078]
实验结果:
[0079]
如下表4所示,测试本发明多拉司琼n-氧化物在10μm浓度下对lyn和mst1激酶活性具有抑制作用。
[0080]
表4:本发明多拉司琼n-氧化物在10μm浓度下对lyn和mst1激酶活性的抑制率
[0081][0082]
上述结果表明,该多拉司琼n-氧化物对lyn和mst1激酶活性抑制的ic
50
值约为10μm,显示了较强的抑制活性。
[0083]
实施例6本发明多拉司琼(组合物1)及甲磺酸多拉司琼(组合物2)的制备
[0084][0085]
在避光及氦气保护下,将10kg丁酮、0.91kg(5mol)内-六氢-8-羟基-2.6-亚甲基-2h-喹嗪-3(4h)-酮及0.8kg(5mol)吲哚-3-甲酸加入反应釜中;搅拌下将1.27kg(10mol)草酰氯缓慢滴加至反应釜中;加毕,将反应液加热至回流反应5小时,tlc检测基本无原料斑点后,加入12kg丁酮与24kg纯化水萃取,分出水层;水层用12kg乙酸乙酯洗涤后,加入固体碳酸钾,调节ph值12~13。过滤,滤饼于60℃下真空干燥5h,得多拉司琼1.23kg(组合物1),收率75.8%。
[0086]
在避光及氦气保护下,将1kg丙酮及123g多拉司琼加入反应釜中,加热回流溶解,滴加入甲烷磺酸,调节体系ph值为2~3。将反应液冷却至20℃,搅拌析晶2小时。过滤,滤饼于55~65℃减压干燥5小时,得甲磺酸多拉司琼135g(组合物2),收率84.8%。
[0087]
实施例7本发明马来酸多拉司琼(组合物3)的制备
[0088]
在避光及氮气保护下,取1kg丙酮及实施例5制得的多拉司琼123g加入反应釜中,加热回流溶解,滴加入马来酸,调节体系ph值为2~3。将反应液冷却至20℃,搅拌析晶3小时。过滤,滤饼于55~65℃减压干燥5小时,得马来酸多拉司琼133g(组合物3),收率82.9%。
[0089]
实施例8本发明磷酸多拉司琼(组合物4)的制备
[0090]
在避光及氮气保护下,取1kg丙酮及实施例5制得的多拉司琼123g加入反应釜中,加热回流溶解,滴加入磷酸,调节体系ph值为2~3。将反应液冷却至20℃,搅拌析晶4小时。过滤,滤饼于55~65℃减压干燥5小时,得磷酸多拉司琼132g(组合物4),收率86.1%。
[0091]
实施例9本发明枸橼酸多拉司琼(组合物5)的制备
[0092]
在避光及氦气保护下,取1kg丙酮及实施例5制得的多拉司琼123g加入反应釜中,加热回流溶解,滴加入枸橼酸,调节体系ph值为2~3。将反应液冷却至20℃,搅拌析晶2小时。过滤,滤饼于55~65℃减压干燥5小时,得枸橼酸多拉司琼160g(组合物5),收率85.3%。
[0093]
实施例10本发明药物组合物的稳定性考察
[0094]
取实施例6制得的多拉司琼(组合物1)、甲磺酸多拉司琼(组合物2)样品及市售的多拉司琼和甲磺酸多拉司琼样品适量,置称量瓶中,厚度约为5mm,分别开口放置在高温(60℃)高湿(25℃、相对湿度(90
±
5)%)、光照(强度为(4500
±
500)lx)环境下考察10d,于第5d及第10d分别取样,观察其外观、色泽均无变化(白色粉末),hplc测定有关物质,与在各环境下考察前(0d)的样品比较,结果见下表5:
[0095]
表5本发明药物组合物与现有产品的稳定性考察结果(%)
[0096]
[0097][0098]
色谱条件:色谱柱:zorbax sb c8反相柱(250
×
4.6mm);流动相a:0.01mol/l磷酸二氢钠缓冲溶液(ph6.8-7.0)-乙腈(1000:53),流动相b:甲醇,梯度洗脱;检测波长:210nm。
[0099]
上表5显示,与市售品比较,本发明组合物1、组合物2多拉司琼n-氧化物含量更低,在高温、高湿、光照条件下其有关物质a和总杂质的增长比市售品要少得多,说明控制本发明多拉司琼n-氧化物的含量有利于组合物中有关物质a和总杂质含量的控制。研究表明,当多拉司琼n-氧化物含量超过0.4%,稳定性考察中总杂质增加更快。
[0100]
实施例11本发明甲磺酸多拉司琼注射液的制备
[0101]
处方:
[0102][0103]
[0104]
制备工艺:
[0105]
称取处方量的甘油,用冷至室温的处方量70%的注射用水完全溶解后,加入处方量的甲磺酸多拉司琼(组合物2)搅拌溶解(浓配),加入浓配体积的0.05%(w/v)的药用活性炭搅拌均匀,静置吸附10min后,过滤脱炭;加注射用水至全量的95%,搅拌均匀,用0.1mol/l盐酸溶液调节ph至3.0,补加注射用水至全量,搅拌均匀;取样测定其性状、ph值、含量和细菌内毒素,合格后精滤、充氮气、灌装、熔封,在121℃湿热灭菌12~15分钟,即得。经高效液相色谱检测,该甲磺酸多拉司琼注射液中甲磺酸多拉司琼与式(ⅰ)化合物之间的质量比为1860:1。
[0106]
实施例12本发明甲磺酸多拉司琼注射液的制备
[0107]
处方:
[0108][0109]
制备工艺:
[0110]
称取处方量的甘露醇,用冷至室温的处方量70%的注射用水完全溶解后,加入处方量的甲磺酸多拉司琼(组合物2)搅拌溶解(浓配),加入浓配体积的0.05%(w/v)的药用活性炭搅拌均匀,静置吸附10min后,过滤脱炭;加注射用水至全量的95%,搅拌均匀,用0.1mol/l盐酸溶液调节ph至3.8,补加注射用水至全量,搅拌均匀;取样测定其性状、ph值、含量和细菌内毒素,合格后精滤、充氮气、灌装、熔封,在121℃湿热灭菌12~15分钟,即得。经高效液相色谱检测,该甲磺酸多拉司琼注射液中甲磺酸多拉司琼与式(ⅰ)化合物之间的质量比为2020:1。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1