一种氨磺必利药典杂质的制备方法与流程
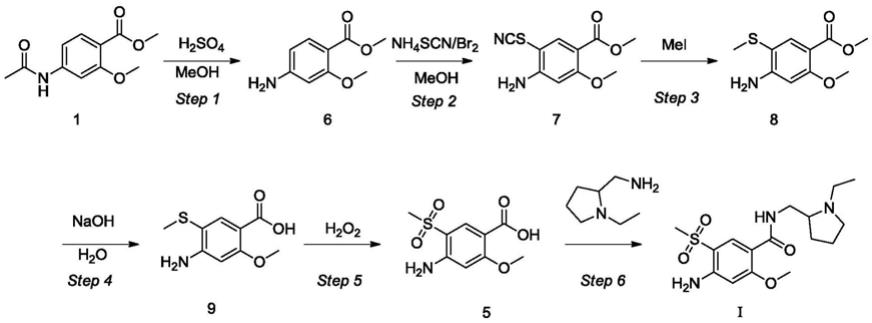
1.本发明属于药物化学技术领域,具体涉及一种氨磺必利药典杂质的制备方法。
背景技术:
2.氨磺必利的化学名为4-氨基-n-[(1-乙基-2-吡咯烷)甲基]-5-乙基磺酰基-2-甲氧基苯甲酰胺,其化学结构式如下:
[0003][0004]
图1:氨磺必利结构式
[0005]
氨磺必利(amisulprid,商品名solian),别名阿米舒必利,是由赛诺菲-圣德拉堡(sanofi-synthelabo)开发的一款多巴胺d3/d2受体拮抗剂。氨磺必利为抗精神病药口服药,临床上主要用于治疗精神分裂症。
[0006]
氨磺必利目前在英国和欧盟均有上市。氨磺必利已被收录在上述二地的药典中。其中,欧洲药典(ep 9.0)在其质量标准中的有关物质检测项中,规定了一个特定杂质d,中文名称为4-氨基-n-[(1-乙基-2-吡咯烷)甲基]-5-甲基磺酰基-2-甲氧基苯甲酰胺。
[0007][0008]
图2:氨磺必利欧洲药典杂质d
[0009]
经过文献查询,发现氨磺必利杂质d的合成方法也有过报道。
[0010][0011]
该合成路线使用了毒性大,环境不友好的碘甲烷和双氧水试剂。碘甲烷是2017年10月27日,世界卫生组织国际癌症研究机构公布的致癌物清单初步整理参考,碘甲烷在3类致癌物清单中。双氧水适用于医用伤口消毒、环境消毒和食品消毒,但过氧化氢也是世界卫
生组织公布的致癌物。而且生产过程中会产生硫酚类中间体,气味特殊恶臭。
技术实现要素:
[0012]
本发明提供一种氨磺必利药典杂质的制备方法,具体为一种氨磺必利药典杂质d的制备方法,该方法操作简单易行、条件温和、收率高、能耗及污染少,适合实验室级别的标准品制备。
[0013][0014]
发明中合成路线中所述的化合物3和化合物4。
[0015]
步骤2所述的催化剂优选自碘化亚铜,氯化亚铜,溴化亚铜或氧化亚铜。
[0016]
步骤3所述的酸选自硫酸,碳酸钾。
[0017]
步骤4所述的碱选自氢氧化钠,氢氧化钾,碳酸铯或碳酸钾。
附图说明
[0018]
图1为式i化合物的1h-nmr谱图。
[0019]
图2为式i化合物的esi-ms谱图。
[0020]
式i所示化合物通过核磁共振(1h nmr)、质谱(esi-ms)进行结构表征,检测结果分别如图1和图2所示。对核磁共振氢谱进行解析,归属如下:
[0021]1h nmr(400mhz,d6-dmso))显示:δ8.18(s,1h,ar),8.06(m,j=3.2hz,1h,conh),8.18(s,1h,ar),6.48(s,2h,nh2),3.88(s,3h,ch3),3.48(m,j=3.2hz,ch),3.15(m,j=3.2hz,ch2),3.07(s,3h,ch3),2.81(m,j=4.4hz,ch),2.58(s,1h,ch),2.23(m,j=5.2hz,ch),2.15(m,j=8.0hz,ch),1.80(m,j=4.0hz,ch),1.65(m,j=7.2hz,ch2),1.50(m,j=3.2hz,ch),1.04(t,j=7.2hz,ch3)。
[0022]
质谱(esi-ms)显示:[m+1]
+
=356.10,式i化合物的理论分子量为355.16。
具体实施方式
[0023]
以下实施例用于说明本发明,应注意,以下描述只是为进一步说明本发明的特征和优点而列举,并不对本发明的权利要求范围进行限制。
[0024]
实施例1:控制温度为15~30℃,将20.0g的化合物1加入70ml乙酸中,慢慢滴加15.3g液溴至反应中,有大量的固体析出,搅拌5-6小时,慢慢滴加50ml正庚烷至反应中,搅
拌1-2小时,过滤,正庚烷漂洗滤饼,滤饼再溶解于300ml二氯甲烷和50ml水中,搅拌分液,有机相再用饱和碳酸钠溶液调节ph=8,搅拌分液,收集二氧甲烷有机相,减压浓缩乙酸乙酯有机相至无液体流出,加入50ml甲基叔丁基醚,搅拌1-2小时,过滤,甲基叔丁基醚漂洗滤饼,湿品于60℃真空烘干后得到25.7g化合物2,黄色固体,收率95%。
[0025]
实施例2:控制温度为15~30℃,将5.0g的化合物1加入17.5ml甲醇中,慢慢滴加3.8g液溴至反应中,有大量的固体析出,搅拌5-6小时,慢慢滴加12.5ml正庚烷至反应中,搅拌1-2小时,过滤,正庚烷漂洗滤饼,滤饼再溶解于75ml二氯甲烷和12.5ml水中,搅拌分液,有机相再用饱和碳酸钠溶液调节ph=8,搅拌分液,收集二氧甲烷有机相,减压浓缩乙酸乙酯有机相至无液体流出,加入12.5ml甲基叔丁基醚,搅拌1-2小时,过滤,甲基叔丁基醚漂洗滤饼,湿品于60℃真空烘干后得到5.9g化合物2,黄色固体,收率90%。
[0026]
实施例3:氮气保护下,将14.0g的化合物2和23.65g甲烷亚磺酸钠,加入70ml二甲基亚砜中,氮气置换空气三次,氮气保护下,加入44g碘化亚铜,升温至90-95℃,搅拌12-16小时,降温至15~30℃。加入100ml水淬灭反应,析出固体,过滤,滤饼用200ml饱和氯化铵溶液和15ml氨水混合溶液漂洗,将湿品加入至100ml饱和氯化铵溶液和15ml氨水混合溶液中,搅拌1-2小时,过滤,滤饼再用水漂洗,湿品于60℃真空烘干后得到11.2g化合物2,黄色固体,收率80%。
[0027]
实施例4:氮气保护下,将14.0g的化合物2和23.65g甲烷亚磺酸钠,加入70ml二甲基亚砜中,氮气置换空气三次,氮气保护下,加入33g溴化亚铜,升温至90-95℃,搅拌12-16小时,降温至15~30℃。加入100ml水淬灭反应,析出固体,过滤,滤饼用200ml饱和氯化铵溶液和15ml氨水混合溶液漂洗,将湿品加入至100ml饱和氯化铵溶液和15ml氨水混合溶液中,搅拌1-2小时,过滤,滤饼再用水漂洗,湿品于60℃真空烘干后得到11.2g化合物3,黄色固体,收率80%。
[0028]
实施例5:将8.0g的化合物3溶解于80ml甲醇中,慢慢滴加7.9g浓硫酸,升温至60-65℃,搅拌1-2小时,降温至15~30℃,生成化合物4。然后将11.7g氢氧化钠溶解于20ml水,然后将氢氧化钠溶液慢慢滴加至反应中,升温至60-65℃,搅拌1-2小时,降温至15~30℃,慢慢滴加2n盐酸溶液调节ph约4左右,黄色固体析出,过滤,滤饼再用水漂洗,湿品于60℃真空烘干后得到5.8g化合物5,黄色固体,收率89%。
[0029]
实施例6:将8.0g的化合物3溶解于80ml甲醇中,慢慢滴加7.9g浓硫酸,升温至60-65℃,搅拌1-2小时,降温至15~30℃,生成化合物4。然后将20.0g碳酸钾溶解于20ml水,然后将溶液慢慢滴加至反应中,升温至60-65℃,搅拌1-2小时,降温至15~30℃,慢慢滴加2n盐酸溶液调节ph约4左右,黄色固体析出,过滤,滤饼再用水漂洗,湿品于60℃真空烘干后得到5.0g化合物5,黄色固体,收率77%。
[0030]
实施例7:将8.0g的化合物3溶解于80ml甲醇中,慢慢滴加11.1g碳酸钾,升温至60-65℃,搅拌1-2小时,降温至15~30℃,生成化合物4。然后将16.2g氢氧化钾溶解于20ml水,然后将溶液慢慢滴加至反应中,升温至60-65℃,搅拌1-2小时,降温至15~30℃,慢慢滴加2n盐酸溶液调节ph约4左右,黄色固体析出,过滤,滤饼再用水漂洗,湿品于60℃真空烘干后得到5.2g化合物5,黄色固体,收率80%。
[0031]
实施例8:将8.0g的化合物3溶解于80ml甲醇中,慢慢滴加7.9g浓硫酸,升温至60-65℃,搅拌1-2小时,降温至15~30℃,生成化合物4。然后将47.1g碳酸铯溶解于20ml水,然
后将溶液慢慢滴加至反应中,升温至60-65℃,搅拌1-2小时,降温至15~30℃,慢慢滴加2n盐酸溶液调节ph约4左右,黄色固体析出,过滤,滤饼再用水漂洗,湿品于60℃真空烘干后得到5.2g化合物5,黄色固体,收率80%。
[0032]
实施例9:将50g的化合物2和250ml丙酮中,慢慢滴加19.5g三乙胺,滴加过程中有溶清过程,继续滴加又有白色固体析出,然后再滴加16.75g氯甲酸乙酯,继续加入29.7g n-乙基-2-氨甲基吡咯烷,保持温度15~30℃,搅拌2-3小时。再滴加19.5g三乙胺和16.75g氯甲酸乙酯,搅拌2-3小时,加入200ml水淬灭反应,用25%氢氧化钠溶液调节ph约12左右析晶,再加入450ml水析晶。过滤后湿品加入250ml丙酮和200ml水中,滴加1n盐酸溶液溶清,用25%氢氧化钠溶液调节ph约12左右析晶,加入15.0g三乙胺,再加入450ml水析晶,过滤,烘料得到白色固体,收率80%。
[0033]
本发明提出的氨磺必利药典杂质d的制备方法已通过实施例进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的氨磺必利药典杂质d的制备方法进行改动或适当的变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明的精神、范围和内容中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1