本发明属于基因检测技术领域,尤其涉及18srna在qrt-pcr检测卵巢癌细胞缺氧刺激前后基因表达中的应用。
背景技术:
卵巢癌死亡率一直高居女性生殖系统恶性肿瘤首位,腹腔内播散和远端转移是其导致死亡的主要原因。因此,卵巢癌细胞的侵袭转移机制一直是妇科研究人员关注的重点和热点。近年来研究发现,上皮间质化(epithelial-mesenchymaltransition,emt)在实体肿瘤的侵袭转移中发挥重要作用。低氧是实体肿瘤微环境的特征之一,在实体肿瘤发生发展过程中普遍存在,参与多种肿瘤生物学的变化。在低氧条件下,实体肿瘤细胞可通过emt促进肿瘤细胞的侵袭和迁移,参与肿瘤的转移。
因此,低氧环境下,为了明确emt在卵巢癌转移过程中的分子机制,对所涉及的emt相关基因定量表达检测在所难免。实时定量pcr(quantitativereal-timepcr,qrt-pcr)就是一种在转录水平对mrna进行定量的手段,具有方便、快捷、重复性好、灵敏度高等优点。因此,qrt-pcr在基因定量表达中获得广泛应用。然而,为了获得qrt-pcr分析的结果,降低实验中的差异,通常在研究中会选择管家基因对目的基因进行校正和标准化,以校正转录效率和cdna用量,弥补制备过程中样本纯度和浓度的差别,从而避免mrna转录效率、样品操作及实验处理步骤造成的误差。
根据实时定量pcr国际化标准指南(miqe)要求,提出了qrt-pcr实验的最少需求,其中就包括合适的内参基因。合适的内参基因指的就是在任何环境条件可稳定表达的管家基因,即该基因的表达不随实验或机体环境的改变而变化。然而,研究表明,机体在不同的环境条件下,多种可作为内参基因的管家基因表达是会随所处环境或处理条件不同而变化的。合适的稳定表达的内参基因对qrt-pcr检测的灵敏度和重复性至关重要。
技术实现要素:
有鉴于此,本发明的目的在于提供18srna在qrt-pcr检测卵巢癌细胞缺氧刺激前后基因表达中的应用;所述18srna在卵巢癌细胞缺氧刺激前后均能稳定表达,作为内参基因为研究卵巢癌细胞缺氧刺激前后基因定量表达水平的数据提供可靠的保障。
为了实现上述发明目的,本发明提供了以下技术方案:
本发明提供了18srna在qrt-pcr检测卵巢癌细胞缺氧刺激前后基因表达中的应用,所述18srna作为qrt-pcr检测的内参基因。
优选的,所述应用包括以下步骤:
1)分别提取缺氧刺激前、缺氧刺激后的卵巢癌细胞rna,反转录后获得缺氧刺激前、缺氧刺激后的卵巢癌细胞的cdna;
2)以步骤1)中所述的缺氧刺激前的卵巢癌细胞的cdna为模板,分别以18srna的引物对和目的基因的引物对为引物进行qrt-pcr扩增,绘制溶解曲线获得缺氧刺激前18srna的表达量和目的基因的表达量,计算获得缺氧刺激前目的基因的相对表达量;
以步骤1)中所述的缺氧刺激后的卵巢癌细胞的cdna为模板,分别以18srna的引物对和目的基因的引物对为引物进行qrt-pcr扩增,绘制溶解曲线获得缺氧刺激后18srna的表达量和目的基因的表达量,计算获得缺氧刺激后目的基因的相对表达量。
优选的,所述qrt-pcr扩增的扩增体系以20μl计,包括以下组分:2×的sybrgreenpremix10μl,10μm的上游引物1.0μl,10μm的下游引物1.0μl,cdna模板2μl和ddh2o6.0μl。
优选的,所述qrt-pcr扩增的扩增程序包括以下步骤:95℃、2min预变性;然后95℃、5s变性,57℃、20s退火,72℃、20s延伸;所述变性-退火-延伸进行40个循环。
优选的,扩增所述18srna的上游引物的核苷酸序列如seqidno.1,扩增所述18srna的下游引物的核苷酸序列如seqidno.2所示。
优选的,所述目的基因包括slug基因和snail基因。
优选的,扩增所述slug基因的上游引物的核苷酸序列如seqidno.3所示,扩增所述slug基因的下游引物的核苷酸序列如seqidno.4所示。
优选的,扩增所述snail基因的上游引物的核苷酸序列如seqidno.5所示,扩增所述snail基因的下游引物的核苷酸序列如seqidno.6所示。
本发明的有益效果:本发明提供的18srna在qrt-pcr检测卵巢癌细胞缺氧刺激前后基因表达中的应用,所述18srna在卵巢癌细胞缺氧刺激前后均能稳定表达,而且能够反应功能基因在不同氧浓度环境下细胞基因的变化,作为内参基因为研究卵巢癌细胞缺氧刺激前后基因定量表达水平的数据提供可靠的保障。
附图说明
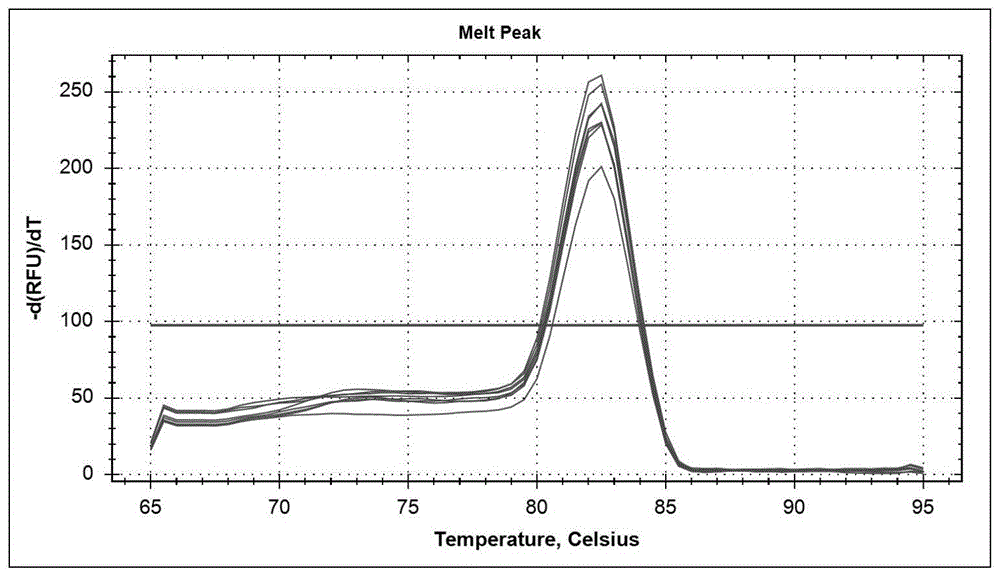
图1为18srna实时荧光定量pcr扩增溶解曲线;
图2为slug基因和snail基因在缺氧刺激前后的相对表达量;
图3为gapdh基因实时荧光定量pcr扩增溶解曲线;
图4为β-actin基因实时荧光定量pcr扩增溶解曲线;
图5为tubb基因实时荧光定量pcr扩增溶解曲线;
图6为ppia基因实时荧光定量pcr扩增溶解曲线;
图7为rpl13a基因实时荧光定量pcr扩增溶解曲线;
图8为tbp基因实时荧光定量pcr扩增溶解曲线;
图9为sdha基因实时荧光定量pcr扩增溶解曲线;
图10为gapdh、β-actin、18srna、tubb、ppia、rpl13a、tbp和sdha表达稳定度比较。
具体实施方式
本发明提供了18srna在qrt-pcr检测卵巢癌细胞缺氧刺激前后基因表达中的应用,所述18srna作为qrt-pcr检测的内参基因;在本发明中,所述18srna优选为人的18srna;所述18srna在ncbi数据库中的序列号为nr_003286.4。
在本发明中,所述应用包括以下步骤:1)分别提取缺氧刺激前、缺氧刺激后的卵巢癌细胞rna,反转录后获得缺氧刺激前、缺氧刺激后的卵巢癌细胞的cdna;2)以步骤1)中所述的缺氧刺激前的卵巢癌细胞的cdna为模板,分别以18srna的引物对和目的基因的引物对为引物进行qrt-pcr扩增,绘制溶解曲线获得缺氧刺激前18srna的表达量和目的基因的表达量,计算获得缺氧刺激前目的基因的相对表达量;以步骤1)中所述的缺氧刺激后的卵巢癌细胞的cdna为模板,分别以18srna的引物对和目的基因的引物对为引物进行qrt-pcr扩增,绘制溶解曲线获得缺氧刺激后18srna的表达量和目的基因的表达量,计算获得缺氧刺激后目的基因的相对表达量。
在本发明中,分别提取缺氧刺激前、缺氧刺激后的卵巢癌细胞rna,反转录后获得缺氧刺激前、缺氧刺激后的卵巢癌细胞的cdna。在本发明中,所述卵巢癌细胞优选为卵巢癌细胞skov3,所述卵巢癌细胞skov3优选的购买于美国atcc。在本发明中,所述缺氧刺激优选的在0.5%~1.5%的o2环境中培养所述卵巢癌细胞20~28h,更优选的在1.0%的o2环境中(94%n2,5%co2和1%o2)培养所述卵巢癌细胞24h。在本发明中,缺氧刺激前所述卵巢癌细胞优选的在常规的21%的o2环境中培养(即常规的空气环境)。本发明对卵巢癌细胞rna的提取方法没有特殊限定,采用本领域常规的rna提取方法即可,具体实施过程参见实施例记载。在本发明中,所述的反转录优选的采用revertrartmastermixwithgdnaremover试剂盒进行,具体操作参见试剂盒说明书。
本发明在获得所述缺氧刺激前、缺氧刺激后的卵巢癌细胞的cdna后,以所述缺氧刺激前的卵巢癌细胞的cdna为模板,分别以18srna的引物对和目的基因的引物对为引物进行qrt-pcr扩增,绘制溶解曲线获得缺氧刺激前18srna的表达量和目的基因的表达量,计算获得缺氧刺激前目的基因的相对表达量。
在本发明中,所述qrt-pcr扩增的扩增体系以20μl计,优选的包括以下组分:2×的sybrgreenpremix10μl,10μm的上游引物1.0μl,10μm的下游引物1.0μl,cdna模板2μl和ddh2o6.0μl;所述qrt-pcr扩增的扩增程序优选的包括以下步骤:95℃、2min预变性;然后95℃、5s变性,57℃、20s退火,72℃、20s延伸;所述变性-退火-延伸进行40个循环。在本发明中,所述绘制溶解曲线的程序优选的如下:温度以1℃/min的速度从60℃递增到95℃,连续测定扩增后样品的荧光强度,获取溶解曲线。在本发明中,所述缺氧刺激前目的基因的相对表达量通过2-△△ct公式计算:其中△ct=目的基因ct-内参基因ct,△△ct=目的基因△ct-内参基因△ct;所述目的基因ct和内参基因ct通过溶解曲线获得。
在本发明中,扩增所述18srna的上游引物的核苷酸序列如seqidno.1,具体如下:ggagcctgcggcttaatttg,扩增所述18srna的下游引物的核苷酸序列如seqidno.2所示,具体如下:ccacccacggaatcgagaaa。
本发明对所述目的基因的种类没有特殊限定;所述卵巢癌细胞中的基因均可作为目的基因;在本发明具体实施过程中,以slug基因和snail基因为例。在本发明中,所述slug基因的ncbi序列号优选为nm_003068.5,所述snailg基因的ncbi序列号优选为nm_005985.4;在本发明中,扩增所述slug基因的上游引物的核苷酸序列如seqidno.3所示,具体如下:ccaagctttcagacccccat;扩增所述slug基因的下游引物的核苷酸序列如seqidno.4所示,具体如下:tgcagctgcttatgtttggc。在本发明中,扩增所述snail基因的上游引物的核苷酸序列如seqidno.5所示,具体如下:accccaatcggaagcctaac;扩增所述snail基因的下游引物的核苷酸序列如seqidno.6所示,具体如下:cagatgagcattggcagcga。
在本发明中,以所述的缺氧刺激后的卵巢癌细胞的cdna为模板,分别以18srna的引物对和目的基因的引物对为引物进行qrt-pcr扩增,绘制溶解曲线获得缺氧刺激后18srna的表达量和目的基因的表达量,计算获得缺氧刺激后目的基因的相对表达量。在本发明中,缺氧刺激后目的基因的相对表达量的获得过程与上述缺氧刺激前目的基因的相对表达量的获得过程,除了cdna为模板替换为缺氧刺激后的卵巢癌细胞的cdna以外,其他步骤和参数均一致,在此不再赘述。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
样品的准备
卵巢癌细胞skov3购买于美国atcc,待细胞长满至6孔板培养皿底面积的80%时,更换无血清培养基继续培养12h后,将细胞分为两组,一组在常规环境下(21%o2)继续培养,另一组给予(1%o2)缺氧培养,24h后收集所有细胞。每孔加入1mltrizol后室温放置5min,然后收集液体于无rna酶的ep管中,放入-80℃备用。
样品rna提取
取出制备好的裂解样品,室温放置5min,使其充分裂解,12000g离心5min,取上清,弃沉淀。按200μl氯仿/(1mltrizol),加入氯仿,轻柔振荡15s,混匀后室温放置3min。4℃,12000g离心15min。取上层无色水相于另一干净无rnase离心管中。按0.5ml异丙醇/(1mltrizol)加入异丙醇轻柔充分混匀,室温放置10min,4℃,12000g离心10min。按1ml75%无水乙醇/(1mltrizol)加入配置好的无水乙醇,洗涤沉淀,4℃,7500g离心5min。室温晾干10min后,用无rnase水50μl溶解rna,55℃,10min水浴。测定浓度后分装,-70℃保存。
反转录
(1)样品前处理:
采用revertrartmastermixwithgdnaremover试剂盒进行cdna合成,反应体系和条件如下:以0.5μgrna为模板,在0.2ml无rnase离心管中于65℃预变性5min,迅速置于冰上冷却。
(2)dna去除:
于低温条件配制下列反应液。向已变性的mrna中加入4×dnmastermix(已添加grnaremover)2μl,然后用nuclease-free水调节反应体系至8μl。将反应液轻轻混匀,在37℃条件下温育5min。
(3)反转录反应
反转录反应,在冰上配制下列反应液。向(2)中反应液中加入5×rtmastermixii反转录酶2μl,使总反应体系到10μl。将反应液轻轻混匀后,按37℃,10min;50℃,5min;98℃,5min使所有酶失活,最后冷却至4℃保存。反应结束后,于-20℃条件下保存。
设计并合成18srna内参基因的引物序列:
上游引物:ggagcctgcggcttaatttg(seqidno.1)
下游引物:ccacccacggaatcgagaaa(seqidno.2)。
引物设计条件为:退火温度60℃,gc含量42~55%,引物长度为19~21bp。
取上述制备的cdna模板10μl进行10倍的倍比稀释,得到浓度为10-2、10-3、10-4、10-5和10-6倍的cdna样品稀释液;样品稀释液作为反应模板进行荧光定量pcr反应,以判断18srna内参基因扩增引物的特异性及其扩增效率。为保证实验结果的可靠性,设置两组平行实验。
实时荧光定量pcr的体系为:sybrgreenpremix(2×)10μl,上游引物(10μm)1.0μl,下游引物(10μm)1.0μl,cdna模板2μl,ddh2o6.0μl,共20μl;
荧光定量pcr扩增反应条件为:95℃、2min预变性,然后变性95℃、5s,退火57℃、20s,延伸72℃,20s。40个循环。实时荧光定量pcr扩增循环结束后分析、绘制溶解曲线,并进行熔解曲线分析以确定扩增产物的特异性。温度以1℃/min的速度从60℃递增到95℃,连续测定扩增后样品的荧光强度,获取溶解曲线。溶解曲线详见图1,由图1可知,18srna内参基因的实时荧光定量pcr扩增溶解曲线均为单峰。因此,说明本发明设计的扩增引物具有特异性。
绘制标准曲线:通过bio-radcfx96系统软件的分析,分别计算以浓度为10-2、10-3、10-4、10-5和10-6倍的cdna样品稀释液作为反应模板的实时荧光定量pcr反应中内参基因18srna的循环阈值即ct值,每个稀释梯度的起始模板浓度与ct值之间呈线性关系,起始模板中所含原始拷贝数越大,ct值越小;根据ct值在excel中经对数拟合做图,得到内参基因引物扩增标准曲线。根据标准曲线斜率确定引物扩增效率y=【10^(-1/slope)-1】,其中y为扩增效率,slope为曲线斜率。本发明中18srna作为内参基因的标准曲线方程、回归系数r2和扩增效率如下:
基因斜率slope扩增效率(%)回归系数r2
18srna-3.35898.50.996
18srna作为检测缺氧刺激高原前后细胞中emt相关基因slug、snail基因的内参基因。
slug基因(nm_003068.5):
上游引物:ccaagctttcagacccccat(seqidno.3)
下游引物:tgcagctgcttatgtttggc(seqidno.4)
snail基因(nm_005985.4):
上游引物:accccaatcggaagcctaac(seqidno.5)
下游引物:cagatgagcattggcagcga(seqidno.6)
实时荧光定量pcr的体系为:sybrgreenpremix(2×)10μl,上游引物(10μm)1.0μl,下游引物(10μm)1.0μl,cdna模板2μl,ddh2o6.0μl,共20μl;
荧光定量pcr扩增反应条件为:95℃、2min预变性,然后变性95℃、5s,退火57℃、20s,延伸72℃,20s。40个循环。实时荧光定量pcr扩增循环结束后分析、绘制溶解曲线,并进行熔解曲线分析以确定扩增产物的特异性。温度以1℃/分钟从60℃缓慢递增到95℃,连续测定样品的荧光强度以获取溶解曲线。利用公式2-△△ct得到基因的相对表达量。其中△ct=目的基因ct-内参基因ct,△△ct=目的基因△ct-对照△ct。
统计分析结果显示,在低氧刺激下,两个目的基因的表达量差异较大,在低氧刺激后,细胞间紧密连接的抑制因子slug和snail表达升高(见图2)。与常氧相比,分别上调了8倍和4倍。因此可以看出,18srna基因在低氧刺激后表达稳定,而且能够反应功能基因在不同氧浓度环境下细胞基因的变化,可作为定量低氧刺激前后卵巢癌细胞中目的基因检测的内参基因。
对比例
分别以gapdh、β-actin、18srna、tubb、ppia、rpl13a、tbp和sdha作为内参基因,进行溶解曲线、扩增效率等的测定。
上述8个基因以及对应的扩增引物见表1。
表1对比内参基因以及引物序列
取实施例1中制备的cdna模板10μl进行10倍的倍比稀释,得到浓度为10-2、10-3、10-4、10-5和10-6倍的cdna样品稀释液;样品稀释液作为反应模板进行荧光定量pcr反应,以判断gapdh、β-actin、18srna、tubb、ppia、rpl13a、tbp和sdha作为内参基因扩增引物的特异性及其扩增效率。为保证实验结果的可靠性,设置两组平行实验。
实时荧光定量pcr的体系为:sybrgreenpremix(2×)10μl,上游引物(10μm)1.0μl,下游引物(10μm)1.0μl,cdna模板2μl,ddh2o6.0μl,共20μl;
荧光定量pcr扩增反应条件为:95℃、2min预变性,然后变性95℃、5s,退火57℃、20s,延伸72℃,20s。40个循环。实时荧光定量pcr扩增循环结束后分析、绘制溶解曲线,并进行熔解曲线分析以确定扩增产物的特异性。温度以1℃/min的速度从60℃递增到95℃,连续测定扩增后样品的荧光强度,获取溶解曲线。溶解曲线详见图1、图3~图7所示,由图1、图3~图7可知,gapdh、β-actin、18srna、tubb、ppia、rpl13a、tbp和sdha作为内参基因的实时荧光定量pcr扩增溶解曲线均为单峰。因此,说明本发明设计的扩增引物具有特异性。
绘制标准曲线:通过bio-radcfx96系统软件的分析,分别计算以浓度为10-2、10-3、10-4、10-5和10-6倍的cdna样品稀释液作为反应模板的实时荧光定量pcr反应中8中内参基因的循环阈值即ct值;每个稀释梯度的起始模板浓度与ct值之间呈线性关系,起始模板中所含原始拷贝数越大,ct值越小;根据ct值在excel中经对数拟合做图,得到内参基因引物扩增标准曲线。根据标准曲线斜率确定引物扩增效率y=10-1/slpoe-1,其中y为扩增效率,slope为曲线斜率。上述8种内参基因的标准曲线方程、回归系数r2和扩增效率如表2所示。
表28种内参基因的标准曲线方程、回归系数r2和扩增效率
用genorm软件程序计算出各个内参基因表达稳定度的平均值m;对各个内参基因的表达稳定度进行排序(m值越小,表达越稳定)。经荧光定量pcr的数据分析结果和genorm软件分析处理后,缺氧刺激前后cdna样品中的gapdh、β-actin、18srna、tubb、ppia、rpl13a、tbp和sdha表达稳定度的平均值m依次为0.016、0.019、0.013、0.017、0.018、0.016、0.015、0.020;表达稳定度由高到低排序依次为18srna=tbp>gapdh>rpl13a>ppia>tubb>β-actin>sdha(见图10)。
由此可见,本发明提供的18srna为稳定性最好的内参基因,以18srna作为参照基因检测缺氧刺激前后卵巢癌细胞不同目的基因的转录表达水平,能够提高对目的基因定量检测的精确度。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>四川大学
<120>18srna在qrt-pcr检测卵巢癌细胞缺氧刺激前后基因表达中的应用
<160>20
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>artificialsequence
<400>1
ggagcctgcggcttaatttg20
<210>2
<211>20
<212>dna
<213>artificialsequence
<400>2
ccacccacggaatcgagaaa20
<210>3
<211>20
<212>dna
<213>artificialsequence
<400>3
ccaagctttcagacccccat20
<210>4
<211>20
<212>dna
<213>artificialsequence
<400>4
tgcagctgcttatgtttggc20
<210>5
<211>20
<212>dna
<213>artificialsequence
<400>5
accccaatcggaagcctaac20
<210>6
<211>20
<212>dna
<213>artificialsequence
<400>6
cagatgagcattggcagcga20
<210>7
<211>20
<212>dna
<213>artificialsequence
<400>7
tccaaaatcaagtggggcga20
<210>8
<211>20
<212>dna
<213>artificialsequence
<400>8
tgatgacccttttggctccc20
<210>9
<211>20
<212>dna
<213>artificialsequence
<400>9
cttccagccttccttcctgg20
<210>10
<211>20
<212>dna
<213>artificialsequence
<400>10
ctgtgttggcgtacaggtct20
<210>11
<211>20
<212>dna
<213>artificialsequence
<400>11
caccttgtctcagccaccat20
<210>12
<211>20
<212>dna
<213>artificialsequence
<400>12
agctcgatactgctggcttc20
<210>13
<211>20
<212>dna
<213>artificialsequence
<400>13
gactgagtggttggatggca20
<210>14
<211>20
<212>dna
<213>artificialsequence
<400>14
tcgagttgtccacagtcagc20
<210>15
<211>20
<212>dna
<213>artificialsequence
<400>15
aaaagcggatggtggttcct20
<210>16
<211>20
<212>dna
<213>artificialsequence
<400>16
gctgtcactgcctggtactt20
<210>17
<211>20
<212>dna
<213>artificialsequence
<400>17
cagcttcggagagttctggg20
<210>18
<211>20
<212>dna
<213>artificialsequence
<400>18
tatattcggcgtttcgggca20
<210>19
<211>20
<212>dna
<213>artificialsequence
<400>19
aaactcgctcttggacctgg20
<210>20
<211>20
<212>dna
<213>artificialsequence
<400>20
tcttccccagcgtttggttt20