一种八氢苯并呋喃衍生物、其制备及应用的制作方法
本发明涉及一种八氢苯并呋喃衍生物、其制备及应用,属于有机合成技术领域。
背景技术:
非甾体抗炎药(non-steroidalanti-inflammatorydrugs,nsaids)具有抗炎、解热、镇痛等作用,在临床上被广泛用于治疗类风湿性关节炎、骨关节炎等疾病,是仅次于抗感染类药物的第二大类药物。该类药物主要通过抑制环氧化酶(cyclooxygenase,cox),阻断花生四烯酸转化为炎症因子前列腺素,从而产生药效。cox包括2种同工酶异构体,即cox1和cox2。
选择性cox2抑制剂是一类新型的nsaids,能选择性地抑制cox2活性,对cox1影响较小,不良反应较少。此外,cox2抑制剂的其他临床用途有抗肿瘤和脑保护作用(皮治兵,等.中国疼痛医学杂志,2006,12(5):297-299.)。因此,对于cox2抑制剂的研究是药物研究中的重要领域。目前,国外一些药厂开发了第二代cox2抑制剂,其主要产品有伐地昔布(valdecoxib)和帕瑞昔布(precoxib),由于伐地昔布和帕瑞昔布均含有磺胺结构,容易引发严重的过敏反应。因此有必要开发一种新的cox2抑制剂来克服现有技术的局限性。
技术实现要素:
针对现有技术中存在的不足,本发明的目的之一在于提供一种八氢苯并呋喃衍生物,即7-硫氰基-3,3-二苯基-八氢苯并呋喃,该物质不含磺胺结构,避免引发过敏反应;
本发明的目的之二在于提供一种八氢苯并呋喃衍生物的制备方法,该方法工艺简单,原料便宜易得,不需要用稀有金属;
本发明的目的之三在于提供一种八氢苯并呋喃衍生物的应用,该物质对于cox2和人卵巢癌细胞具有抑制作用,在生物医药领域具有良好的应用前景。
本发明的目的是通过以下技术方案实现的。
一种八氢苯并呋喃衍生物,所述衍生物为7-硫氰基-3,3-二苯基-八氢苯并呋喃,具体结构式如下所示:
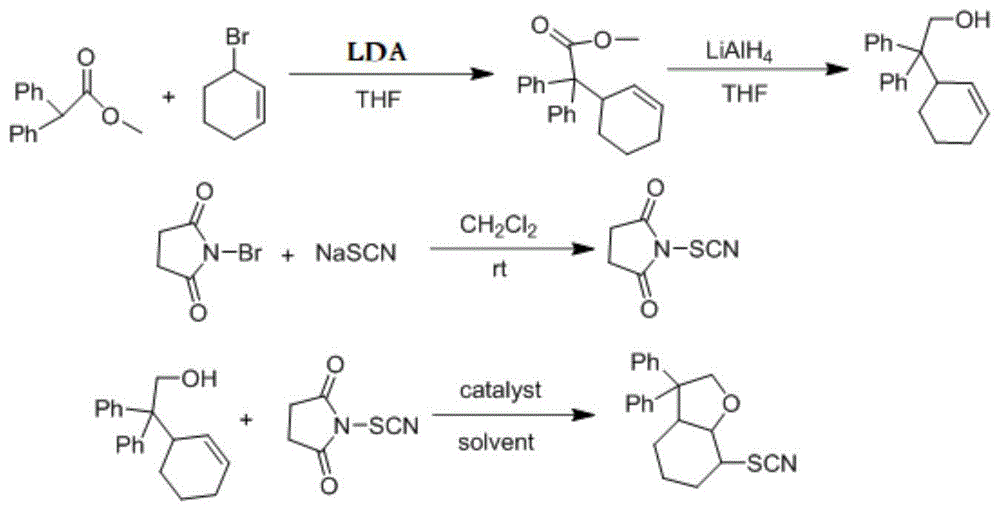
本发明所述八氢苯并呋喃衍生物的制备方法,所述方法步骤如下:利用二苯基乙酸甲酯和3-溴环己烯反应得到2-(环己-2-烯基)-2,2-二苯基乙酸甲酯,2-(环己-2-烯基)-2,2-二苯基乙酸甲酯再与氢化铝锂反应得到2-(环己-2-烯基)-2,2-二苯基乙醇;利用n-溴代琥珀酰亚胺与硫氰酸钠反应得到n-硫氰基琥珀酰亚胺(nts);在无水无氧的条件下以及复合催化剂的作用下,2-(环己-2-烯基)-2,2-二苯基乙醇与n-硫氰基琥珀酰亚胺在有机溶剂中反应不少于8h,再经过硅胶柱纯化得到7-硫氰基-3,3-二苯基-八氢苯并呋喃。
其中,复合催化剂是由三氟甲磺酸盐与膦配体按照0.1:(0.1~0.2)的摩尔比复配形成的,三氟甲磺酸盐优选三氟甲磺酸铜(cu(otf)2)或三氟甲磺酸锌(zn(otf)2),膦配体优选(±)-2,2'-双-(二苯膦基)-1,1'-联萘(binap)或n,n'-双(二苯基膦)联萘胺,三氟甲磺酸盐与膦配体的摩尔比优选0.1:0.12;有机溶剂优选二氯甲烷、乙腈、四氢呋喃或丙酮;2-(环己-2-烯基)-2,2-二苯基乙醇与n-硫氰基琥珀酰亚胺的摩尔比优选1:2。
2-(环己-2-烯基)-2,2-二苯基乙醇制备的具体条件如下:以四氢呋喃为溶剂,以二异丙基氨基锂为催化剂,二苯基乙酸甲酯与3-溴环己烯按照1:(1~1.5)的摩尔比在无水无氧的条件下反应不少于20h,然后淬灭反应,再经过洗涤、干燥、纯化得到2-(环己-2-烯基)-2,2-二苯基乙酸甲酯;以四氢呋喃为溶剂,2-(环己-2-烯基)-2,2-二苯基乙酸甲酯与氢化铝锂按照1:(1~1.5)的摩尔比在无水无氧的条件下反应不少于20h,然后按照h2o、naoh、h2o的顺序进行淬灭反应,且h2o:naoh:h2o的体积比为1:2:3,再经过洗涤、干燥、纯化得到2-(环己-2-烯基)-2,2-二苯基乙醇。
n-硫氰基琥珀酰亚胺制备的具体条件如下:以二氯甲烷为溶剂,n-溴代琥珀酰亚胺与硫氰酸钠按照1:1.5的摩尔比在在无水无氧以及避光的条件下反应1h~3h,先用硅藻土过滤,再避光减压浓缩得n-硫氰基琥珀酰亚胺。
本发明所述八氢苯并呋喃衍生物在制备cox2抑制剂中的应用。
本发明所述八氢苯并呋喃衍生物在制备抑制人卵巢癌细胞药物中的应用。
有益效果:
本发明所述的八氢苯并呋喃衍生物不含有磺胺结构,对于cox2和人卵巢癌细胞具有很好的抑制作用,而且不会引发过敏反应;另外,该衍生物的制备工艺简单,原料便宜易得,不需要用稀有金属,在生物医药领域具有良好的应用前景。
附图说明
图1为本发明所述7-硫氰基-3,3-二苯基-八氢苯并呋喃的合成路线示意图。
图2为实施例5中在添加以及未添加7-硫氰基-3,3-二苯基-八氢苯并呋喃的条件下培养细胞的结果对比图。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步阐述,其中,所述方法如无特别说明均为常规方法,所述原材料如无特别说明均能从公开商业途径而得。
实施例1
7-硫氰基-3,3-二苯基-八氢苯并呋喃的具体制备步骤如下:
(1)取250ml双口圆底烧瓶、磁子置于磁力搅拌器上,通过氩气置换保证烧瓶内无水无氧;先加入无水无氧四氢呋喃(thf)(4ml),然后加入二异丙胺溶液(0.40ml,2.87mmol,1.3equiv),再在零下78℃加入n-buli(正丁基锂,1.65ml,1.6mol/l,2.65mmol,1.2equiv)原位生成二异丙基氨基锂(lda),继续搅拌30min后,再将二苯基乙酸甲酯(500mg,2.21mmol)的无水无氧thf(5ml)溶液在零下78℃下1h内缓慢滴加到烧瓶中,搅拌1h,再加入3-溴环己烯(0.28ml,2.43mmol,1.1equiv),使烧瓶的反应体系在自然条件下升温至室温后搅拌反应24h,通过薄层色谱(tlc)检测反应完成;在搅拌条件下,用5ml盐酸(1mol/l)和10ml蒸馏水淬灭反应,分液取上层有机相,再依次用二氯甲烷洗涤、饱和食盐水去水、na2so4干燥、减压浓缩、硅胶柱色谱法纯化(洗脱剂是体积比为1:49的乙酸乙酯与石油醚),得到透明无色油状物2-(环己-2-烯基)-2,2-二苯基乙酸甲酯,产率为92%;
(2)取250ml双口圆底烧瓶、磁子置于磁力搅拌器上,通过氩气置换保证烧瓶内无水无氧;先加入lialh4(96mg,2.52mmol,1.4equiv)的thf(2.6ml)悬浮液,再在0℃下,缓慢滴加2-(环己-2-烯基)-2,2-二苯基乙酸甲酯(552mg,1.80mmol)的thf(2ml)溶液,使烧瓶的反应体系在自然条件下升至室温后搅拌反应24h,通过薄层色谱(tlc)检测反应完成;在搅拌条件下,用h2o(2.5ml)、naoh(5.0ml,1.0mol/l)、h2o(7.5ml)进行淬灭,先通过硅藻土过滤收集固体产物,再依次用乙酸乙酯洗涤、饱和食盐水去水、无水硫酸钠干燥、减压浓缩、硅胶柱色谱法纯化(洗脱剂是体积比为1:20的乙酸乙酯与石油醚),得白色固体颗粒2-(环己-2-烯基)-2,2-二苯基乙醇,产率为73%;
(3)取250ml双口圆底烧瓶、添加磁子置于磁力搅拌器上并且避光,通过氩气置换保证烧瓶内无水无氧;n-溴代琥珀酰亚胺(nbs)(6mmol)、nascn(9mmol)和无水无氧ch2cl2(45ml)的混合体系在烧瓶中搅拌反应2h后,先用硅藻土过滤除去nabr,再在30℃下蒸发溶剂,在溶剂蒸发到三分之二时,往旋蒸瓶中滴加正己烷,当出现白色固体时,再滴入少量正己烷,继续减压浓缩,得到纯白色固体nts,产率为83%;
(4)取25ml的反应管并加入磁子,加入zn(otf)2(0.1equiv)和binap(0.12equiv)后置于磁力搅拌器上,通过氩气置换保证反应管内无水无氧,之后加入0.5ml无水无氧ch3cn,搅拌0.5h后,再在氩气保护下加入2-(环己-2-烯基)-2,2-二苯基乙醇(0.05mmol)和n-硫氰基琥珀酰亚胺(1.6equiv)进行反应29h,通过硅胶柱色谱法纯化(洗脱剂是体积比为1:20的乙酸乙酯与石油醚),得透明无色油状物7-硫氰基-3,3-二苯基-八氢苯并呋喃,产率66%,合成路线详见图1。
对所制备的7-硫氰基-3,3-二苯基-八氢苯并呋喃进行核磁共振氢谱、核磁共振碳谱、红外光谱以及高分辨质谱表征,表征结果如下:
1h-nmr(400mhz,cdcl3):δ(ppm)=7.38–7.10(m,10h),4.78(d,j=9.0hz,1h),4.28(m,2h),3.41–3.33(m,1h),3.29(m,1h),2.02–1.39(m,6h);
13c-nmr(100mhz,d6-dmso)δ(ppm)=146.2,143.8,129.0,128.7,128.6,127.2,126.8,126.6,112.3,79.9,75.5,58.7,48.7,42.4,26.4,23.7,19.9;
ir(kbr):ν(cm-1)=2150,1597,1493,1447,1067;
hrms(esi)m/z:[m+nh4]+calculatedforc21h21nosnh4353.1682;found353.1679。
实施例2
步骤(1)~(3)与实施例1相同;
(4)取25ml的反应管并加入磁子,加入zn(otf)2(0.1equiv)和binap(0.12equiv)后置于磁力搅拌器上,通过氩气置换保证反应管内无水无氧,之后加入0.5ml无水无氧ch2cl2,搅拌0.5h后,再在氩气保护下加入2-(环己-2-烯基)-2,2-二苯基乙醇(0.05mmol)和n-硫氰基琥珀酰亚胺(1.6equiv)进行反应29h,通过硅胶柱色谱法纯化(洗脱剂是体积比为1:20的乙酸乙酯与石油醚),得透明无色油状物7-硫氰基-3,3-二苯基-八氢苯并呋喃,产率为79%。
实施例3
步骤(1)~(3)与实施例1相同;
(4)取25ml的反应管并加入磁子,加入zn(otf)2(0.1equiv)和binap(0.12equiv)后置于磁力搅拌器上,通过氩气置换保证反应管内无水无氧,之后加入0.5ml无水无氧ch2cl2,搅拌0.5h后,再在氩气保护下加入2-(环己-2-烯基)-2,2-二苯基乙醇(0.05mmol)和n-硫氰基琥珀酰亚胺(2.0equiv)进行反应29h,通过硅胶柱色谱法纯化(洗脱剂是体积比为1:20的乙酸乙酯与石油醚),得透明无色油状物7-硫氰基-3,3-二苯基-八氢苯并呋喃,产率为84%。
实施例4
步骤(1)~(3)与实施例1相同;
(4)取25ml的反应管并加入磁子,加入cu(otf)2(0.1equiv)和n,n'-双(二苯基膦)联萘胺(0.12equiv)后置于磁力搅拌器上,通过氩气置换保证反应管内无水无氧,之后加入0.5ml无水无氧乙腈,搅拌0.5h后,再在氩气保护下加入2-(环己-2-烯基)-2,2-二苯基乙醇(0.05mmol)和n-硫氰基琥珀酰亚胺(2.0equiv)进行反应29h,通过硅胶柱色谱法纯化(洗脱剂是体积比1:20的乙酸乙酯与石油醚),得透明无色油状物7-硫氰基-3,3-二苯基-八氢苯并呋喃,产率58%。
实施例5
将小鼠单核巨噬细胞raw264.7铺种于6孔培养板,加入培养液,于37℃、co2体积分数为5%的培养箱常规培养24h,之后加入脂多糖(lps)使其终浓度为5μg/ml刺激巨噬细胞4h,再加入7-硫氰基-3,3-二苯基-八氢苯并呋喃(p0105)使其终浓度为10μmol/l,然后在37℃、5%co2培养箱中继续孵育24h,弃上清,加入含蛋白酶抑制剂的放射免疫沉淀法缓冲液(ripa)裂解液,在冰上裂解后用细胞刮轻轻刮下,12000r/min、4℃离心5min,取上清液,按照bca(bicinchoninicacid)蛋白浓度测定法测定蛋白含量;之后按照westernblot法检测细胞内cox2的蛋白含量;实验重复三次,并进行统计学分析,结果如图2所示。其中,con为正常对照组(与p0105实验组相比,未用脂多糖刺激也未添加7-硫氰基-3,3-二苯基-八氢苯并呋喃,培养箱中常规培养48h的raw264.7细胞),mod为模型对照组(与p0105实验组相比,只是将7-硫氰基-3,3-二苯基-八氢苯并呋喃替换成等体积的溶媒二甲基亚砜,其他条件不变)。
图2的表征结果表明:终浓度为10μmol/l的7-硫氰基-3,3-二苯基-八氢苯并呋喃对cox2具有非常显著的抑制作用。
实施例6
将对数生长期的肿瘤细胞a2780、sk-ov-3、kuramochi、ovcar8分别用胰酶消化后配制成浓度为2.5×104细胞/ml的细胞液,按2500个/孔接种于96孔板,每孔加100μl;次日换新鲜培养基,设6组实验组且每组设3个平行孔,6个实验组中分别加入7-硫氰基-3,3-二苯基-八氢苯并呋喃使其终浓度依次为0.001、0.01、0.1、1、10和100μmol/l,设1组空白对照组且包含3个平行孔,空白对照组中加入100μl溶媒二甲基亚砜(dmso终浓度<0.5%),设6组阳性对照组且每组设3个平行孔,6个阳性对照组中分别加入紫杉醇的二甲基亚砜溶液使紫杉醇的终浓度依次为0.001、0.01、0.1、1、10和100μmol/l,所有测试组于37℃继续培养72h后,每孔加入20μl新鲜配制的含5mg/ml3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(mtt)的磷酸缓冲盐溶液,继续培养4h,弃上清后,每孔加入150μldmso使mtt甲簪沉淀溶解,微型振荡器振荡混匀后,在参考波长450nm,检测波长570nm条件下测定光密度值(od),以溶剂处理的肿瘤细胞为对照组用下面公式计算受试化合物对肿瘤细胞的生长抑制率,并按中效方程计算半数抑制率(ic50)值。结果表明:7-硫氰基-3,3-二苯基-八氢苯并呋喃对人卵巢癌细胞a2780、sk-ov-3、kuramochi、ovcar8的ic50(μmol/l)依次为42.38、72.71、89.21、97.55。
综上所述,以上仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!