一种应用于双链DNA片段的纯化方法与流程
本发明涉及生物化学与分子生物学技术领域,尤其涉及一种应用于双链dna片段的纯化方法。
背景技术:
质粒(plasmid)广泛存在于生物界,从细菌、放线菌、丝状真菌、大型真菌、酵母到植物,甚至人类机体中都含有。细菌质粒是细菌、酵母菌和放线菌等生物中染色体(或拟核)以外的闭合环状的双链dna分子,大小可为1kb到200kb,具有自主复制能力,并能稳定表达所携带的遗传信息,是基因工程中最常用的载体。
目前已经有技术对分子量相差较大的质粒进行分离的工艺较为成熟,但是很少有对酶切之后的双链dna片段进行分离纯化工艺探索与优化,尤其是对于酶切之后两个片段相差3000bp之内的质粒进行分离纯化,因而具有很好的探索及应用空间,以有利于后期的基因治疗领域的发展。
技术实现要素:
本发明的目的是要建立一套能够简易完成分离纯化酶切之后分子量相差3000bp之内的双链线性dna分子的方法,以有利于后期的基因治疗领域的发展。
为了实现上述目的,本发明采用了如下技术方案:
一种应用于双链dna片段的纯化方法,该方法通过一步层析完成,所用原料及运作参数如下:
层析系统:aktapure150;
层析柱尺寸:21.2*150mm;
流速:3~6ml/min;
流动相a-平衡缓冲液、流动相b-上样洗脱液;
平衡:至基线平稳;
上样:80ml酶切产物;
wash:2~4个cv;
线性洗脱:28%-83%(流动相b)洗25cv。
本发明公布的技术方案可以通过工艺路径的方法将经酶切之后片段进行分离纯化,并且可完成工艺放大,同时达到gmp的条件。
进一步的,所述层析柱所用填料为gesource30q,该骨架采用聚苯乙烯,应用过程中流速高,稳定性好,其活性基团:r-o-ch2-choh-ch2-o-ch2-choh-ch2-n+(ch3)3,可以有效结合带负电荷的基团,平均粒径较小为30μm,分辨率较好。
进一步的,所述基线平稳具体至紫外值固定变化在0.5-1mau。
进一步的,所述平衡缓冲液:ph6.5,50mmtris-hcl,1mmedta;所述上样洗脱液:ph6.5,50mmtris-hcl,1mmedta,2mnacl。
进一步的,所述缓冲液ph值较高,并形成比核酸等电点ph高的环境,使得核酸带净负电荷,可被填料吸附。不同长度的核酸带电量不同,吸附强度不一,依次解离纯化分离。
进一步的,所述酶切产物为分子量相差3000bp之内的双链线性dna分子。
更进一步的,所述酶切产物为分子量相差500~3000bp之内的双链线性dna分子。
进一步的,所述酶切产物经过超滤浓缩无菌化处理,并以pvdf滤膜去除内毒素。
进一步的,所述层析为阴离子交换层析,吸附核酸于填料上,经过洗脱,得到目的片段。
进一步的,所述层析过程后进行误差线性曲线的绘制,包括如下步骤:
先用nanodrop2000测定该双链dna片段浓度,然后用tebuffer稀释至10μg/ml,20μg/ml,50μg/ml,200μg/ml,500μg/ml,1000μg/ml五个浓度梯度,量取峰面积做线性曲线,所用原料及试剂如下:
层析柱:tskgeldna-npr色谱柱;
试剂:流动相a-平衡缓冲液(20mmtris-hcl,ph9.0);
流动相b-洗脱缓冲液(20mmtris-hcl,1mnacl,ph9.0);
0-100%洗脱;
流速:1ml/min;
线性洗脱:0→55%b,22cv,55%-65%b,20cv。
更进一步的,所述洗脱过程为通过核酸吸附强度的差异性,致使较小长度的核酸先脱落。
更进一步的,所述wash:4个cv;所述线性洗脱:32%-78%(流动相b)洗26cv。
综上所述,由于采用了上述技术方案,本发明的有益效果是:
本发明根据核酸分子在中性以及ph偏大情况下带负电荷的特性,采用阴离子交换层析,把核酸吸附在填料上,经过洗脱,即可得到目的片段;本发明可以通过工艺路径的方法将经酶切之后片段的分离纯化,并且可进行工艺放大,可以达到gmp的条件,核酸等电点在2-4左右,在ph较高的环境中,核酸带净负电荷,可以被填料吸附,不同长度的核酸,核酸带电量不同,从而对填料的吸附能力不同,带电量大的结合的牢固,需要较高的电荷置换量才能洗脱掉,所以不同长度的核酸片段会先后被洗脱,达到分离效果,有利于整个行业的发展,本发明的工艺简单有效,一步层析就可以达到效果。
附图说明
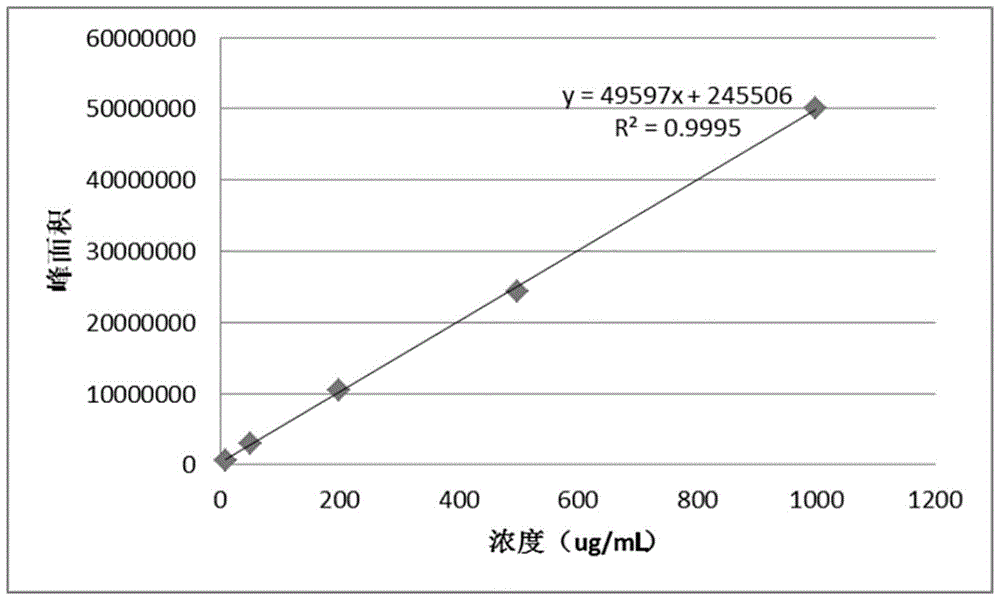
图1是实施例1的质粒浓度-峰面积误差线性曲线的制作;
图2是现有常规方法对分子量相差较大质粒分离的层析谱图;
图3为该图2的分开质粒的凝胶图谱;
图4是现有工艺质粒酶切后进行纯化分离所得谱图;
图5是图4的凝胶电泳图;
图6是本发明方案对酶切后质粒分离试验的层析谱图;
图7是图6经复测所得层析谱图;
图8是图6的凝胶电泳图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
纯化方法的层析过程后进行误差线性曲线的绘制,包括如下步骤:
先用nanodrop2000测定该双链dna片段浓度,然后用tebuffer稀释至10μg/ml,20μg/ml,50μg/ml,200μg/ml,500μg/ml,1000μg/ml五个浓度梯度,量取峰面积做线性曲线,所用原料及试剂如下:
层析柱:tskgeldna-npr色谱柱;
试剂:流动相a-平衡缓冲液(20mmtris-hcl,ph9.0);
流动相b-洗脱缓冲液(20mmtris-hcl,1mnacl,ph9.0);
0-100%洗脱;
流速:1ml/min;
线性洗脱:0→55%b,22cv,55%-65%b,20cv。
一种应用于双链dna片段的纯化方法,该方法包括如下:
对分子量相差3000bp之内的双链线性dna分子进行阴离子交换层析,所用材料及运作参数如下:
层析系统:aktapure150;
层析柱:gesource30q(21.2*150mm);
流速:3~6ml/min;
流动相a-平衡缓冲液(ph6.550mmtris-hcl,1mmedta);
流动相b-上样洗脱液(ph6.550mmtris-hcl,1mmedta,2mnacl);
平衡:至基线平稳(至紫外值固定变化在0.5-1mau);
上样:80ml酶切产物;
wash:3个cv;
线性洗脱:28%-83%(流动相b)洗25cv。
所用器皿经干烤除热源,除菌处理。
实施例1
先用nanodrop2000测定该双链dna片段浓度,然后用tebuffer稀释至10μg/ml,20μg/ml,50μg/ml,200μg/ml,500μg/ml,1000μg/ml五个浓度梯度,量取峰面积做线性曲线,所用原料及试剂如下:
层析柱:tskgeldna-npr色谱柱;
试剂:流动相a-平衡缓冲液(20mmtris-hcl,ph9.0);
流动相b-洗脱缓冲液(20mmtris-hcl,1mnacl,ph9.0);
0-100%洗脱;
流速:1ml/min;
线性洗脱:0→55%b,22cv,55%-65%b,20cv。
所得线性曲线如图1所示。
一种应用于双链dna片段的纯化方法,该方法通过一步层析完成,所用原料及运作参数如下:
具体的,层析系统:aktapure150;
具体的,层析柱尺寸:21.2*150mm;
具体的,流速:3~6ml/min;
具体的,流动相a-平衡缓冲液、流动相b-上样洗脱液;
具体的,平衡:至基线平稳;
具体的,上样:80ml酶切产物;
具体的,wash:2~4个cv;
具体的,线性洗脱:28%-83%(流动相b)洗26cv。
其中,所述层析柱所用填料为gesource30q,应用过程中流速高,稳定性好,其活性基团:r-o-ch2-choh-ch2-o-ch2-choh-ch2-n+(ch3)3可以有效结合带负电荷的基团,平均粒径较小为30μm,分辨率较好。
其中,所述基线平稳具体至紫外值固定变化在0.5-1mau。
其中,所述平衡缓冲液:ph6.5,50mmtris-hcl,1mmedta;所述上样洗脱液:ph6.5,50mmtris-hcl,1mmedta,2mnacl。
其中,所述缓冲液ph值较高,并形成比核酸等电点ph高的环境,使得核酸带净负电荷,可被填料吸附。不同长度的核酸带电量不同,吸附强度不一,依次解离纯化分离。
其中,所述酶切产物为分子量相差3000bp之内的双链线性dna分子。
其中,所述酶切产物经过超滤浓缩无菌化处理,并以pvdf滤膜去除内毒素。
其中,所述层析为阴离子交换层析,吸附核酸于填料上,经过洗脱,得到目的片段。
其中,所述洗脱过程为通过核酸吸附强度的差异性,致使较小长度的核酸先脱落。
其中,所述wash:4个cv;所述线性洗脱:32%-78%(流动相b)洗26cv。
所用器皿经干烤除热源,除菌处理。
对于上述纯化方法所得结果的检核过程,包括如下步骤:
(1)预实验:酶切体系验证1μgdna,10units的酶切体系酶切效果,如果效果达到预期则进行后续步骤:
s1.初步酶切1mg质粒;
s2平衡:用平衡缓冲液平衡层析柱gesource30q;
s3.上样:上样1mg质粒;
s4.wash:平衡缓冲液平衡2cv的柱体积;
s5.洗脱:验证51%,52%,53%,54%,55%的梯度洗脱;
s6.收样:收集两个洗脱峰;
s7.检测:凝胶检测大小是否复合预期,纯度是否复合预期;
所用平衡缓冲液:ph8.010mmtris-hcl,1mmedta,0.4mnacl;
所用洗脱缓冲液:ph8.010mmtris-hcl,1mmedta,1mnacl;
灭菌:试剂瓶干烤除热源。
后续所用溶液与预实验溶液为同一配制批次。
(2)放大上样:
s1.酶切5mg质粒上样:
以51%,52%,53%,54%,55%的梯度洗脱验证酶切效果,如果效果符合预期,则进行后续步骤试验,如果不能符合预期则采用线性梯度洗脱50-60%,5cv的方式收样;
s2.酶切10mg质粒上样:
以51%,52%,53%,54%,55%的梯度洗脱验证酶切效果,如果效果符合预期,则进行后续步骤试验;
s3.酶切20mg质粒上样:
以51%,52%,53%,54%,55%的梯度洗脱验证酶切效果,如果效果符合预期,则进行后续步骤试验;
根据收养多少决定是否继续纯化,之后再下接如下步骤;
(3)超滤浓缩:
将多次纯化后样品用10kd的50ml离心管8000prm/min离心10min超滤换液;
要求:去除内毒素(科晶)、0.22μm过滤器除菌(科晶)。
(3)纯度检测,原料及参数如下:
色谱柱:tskgeldna-npr;
流速:1ml/min;
检测:254nm波长;
流动相a-平衡缓冲液(20mmtris-hcl,ph9.0);
流动相b-洗脱缓冲液(20mmtris-hcl,1mnacl,ph9.0);
上样:100μl,(酶切产物:a=1:1);
线性:0-55%,22cv,55→65%b,20cv;
(4)浓度检测,原料及参数如下:
色谱柱:tskgeldna-npr;
流速:1ml/min;
检测:254nm波长;
流动相a-平衡缓冲液(20mmtris-hcl,ph9.0);
流动相b-洗脱缓冲液(20mmtris-hcl,1mnacl,ph9.0);
上样:50μl,(酶切产物:a=1:1);
线性:50→65%b,10cv;
(5)凝胶检测:配制1%的凝胶检测纯度;
(6)内毒素检测:移交qc检测;
(7)无菌检测:移交qc检测。
所得结果符合分离纯化要求。
用现有的纯化方法和本发明提供的纯化方法进行相关试验,所得谱图及结果如下:
请参阅图1,确定误差曲线,标注了核酸片段的浓度定量关系,根据该图谱,上样后计算峰面积,带入公式可计算核酸浓度;
请参阅图2,现有工艺条件下不同大小的质粒的分离效果,可对不同分子量的质粒进行分离;
请参阅图3,为该图2的质粒分离的凝胶图谱,其中,1号样品为两个待纯化的混合质粒,2好样品为分子量较大的质粒对照凝胶电泳条带,3为纯化后的分子量较大的质粒,4为纯化后分子量较小的质粒,5为分子量较小的质粒对照凝胶结果。结果显示,可以进行不同分子量的质粒纯化分离;
请参阅图4,为现有工艺质粒酶切后进行纯化分离所得图谱,由该图谱可知,出现一个吸收峰,说明没有完成对片段的有效分离,所得效果较差;
请参阅图5,为图4的凝胶结果,maker后左起第一条带是质粒对照,第二条带是纯化后的收集峰凝胶结果,根据条带位置,能够直观的判断条带没有分开;
由图2~5,可知现有技术主要是针对较大质粒的进行分离,所得效果较为显著,可以达到较好的效果,但对较小的如分离酶切后的质粒的效果差。
请参阅图6,通过本发明提供的技术方案,质粒酶切之后进行纯化,纯化峰分为两个,分别为分子量较小的片段和分子量较大的片段,分离效果较好;
请参阅图7,采用与图6所用的相同方案重复一遍,分离效果较好,重现性也较好,表明本发明提供的纯化方法稳定有效;
请参阅图8,将图六中纯化后收集的两个目的产物进行凝胶电泳,第一个是进行质粒酶切之后的电泳结果,后两个是酶切之后纯化的片段,分为大小两个片段。
综上所述,按照本发明公布的方法进行纯化,可以很好的分离小于2000bp的核酸片段。
区别于现有技术:本发明根据核酸分子在中性以及ph偏大情况下带负电荷的特性,采用阴离子交换层析,把核酸吸附在填料上,经过洗脱,即可得到目的片段;本发明可以通过工艺路径的方法进行酶切之后片段的分离纯化,并且可进行工艺放大,可以达到gmp的条件,有利于整个行业的发展,本发明的工艺简单有效,一步层析就可以达到效果。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!