一种2-氯-3-三氟甲基吡啶合成装置的制作方法
本实用新型涉及化学合成技术领域,具体涉及一种2-氯-3-三氟甲基吡啶合成装置。
背景技术:
2-氯-3-三氟甲基吡啶是一种新型的农药中间体,在有机合成染料、医药等领域具有广泛的用途,也是合成高效低毒的除草剂“吡嘧磺隆”的关键中间体。
现有技术中,一般以2-氯-3-甲基吡啶为原料,在催化剂的存在下合成2-氯-3-三氟甲基吡啶,合成的路线大致有两条:1)在反应器中氯氟化一步合成2-氯-3-三氟甲基吡啶;2)分步法,先氯化然后氟化合成2-氯-3-三氟甲基吡啶,现有的技术中的以上两种常见合成方法合成2-氯-3-三氟甲基吡啶的反应一般均是在催化剂的催化作用下进行的。然而所用的催化剂或者价格昂贵(如氟化锰等)、或者不易获得(如氯化钴等)、或者不安全,极易爆炸(如偶氮类);现有的2-氯-3-三氟甲基吡啶的合成设备浪费氯气现象严重,使得工业合成生产成本提高,而且存在严重的安全隐患。
技术实现要素:
本实用新型所要解决的技术问题在于现有技术中2-氯-3-三氟甲基吡啶的合成过程需要使用到易爆炸、昂贵或不易获得的催化剂以及氯气浪费严重等相关问题。本实用新型所述2-氯-3-三氟甲基吡啶的合成装置能够实现不用催化剂即可制备高纯度2-氯-3-三氟甲基吡啶产品的需求,合成路线简单高效,原料成本低,所得产品纯度高,大幅度提升生产安全性,易于工业化推广。
为解决上述技术问题,本实用新型采用的技术方案如下:
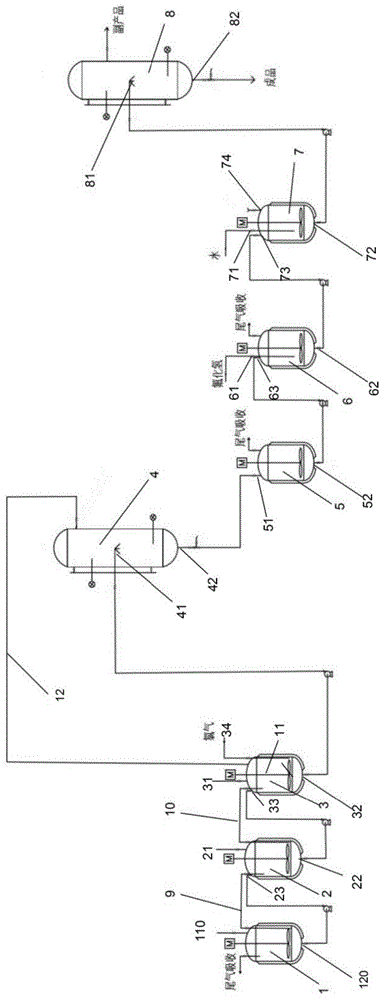
一种2-氯-3-三氟甲基吡啶合成装置,包括依次连通设置的多级氯化釜、第一精馏装置、结晶釜、氟化釜、中和釜和第二精馏装置;所述多级氯化釜包括两级以上的氯化釜。
所述多级氯化釜包括一级氯化釜、二级氯化釜、三级氯化釜;
所述三级氯化釜上端设置有第一进料口,下端设置有第一出料口;
所述二级氯化釜上端设置有第二进料口和第一倒料口,下端设置有第二出料口;
所述一级氯化釜上端设置有第三进料口、第二倒料口和氯气导入口,下端设置有第三出料口;
所述三级氯化釜和二级氯化釜之间设置有第一氯气通道,所述二级氯化釜和一级氯化釜之间设置有第二氯气通道;
所述第一出料口与第一倒料口连通设置,所述第二出料口和第二倒料口连通设置。
进一步地,所述第一精馏装置设置有第四进料口和第四出料口;所述第四进料口与第三出料口连通设置。
进一步地,所述结晶釜设置有第五进料口和第五出料口;所述第五进料口与第四出料口连通设置。
进一步地,所述氟化釜设置有氟化氢导入口、第六出料口和第六进料口;所述第六进料口与第五出料口连通设置。
进一步地,所述中和釜设置有水导入口、第七出料口、第七进料口和碱导入口;所述第七进料口与第六出料口连通设置。
进一步的,所述第二精馏装置设置有第八进料口和成品出料口;所述第八进料口与第七出料口连通设置。
进一步地,所述三级氯化釜、二级氯化釜、一级氯化釜、结晶釜、氟化釜和中和釜中均设置有搅拌装置。
进一步地,所述一级氯化釜和第一精馏装置之间设置有回流管道。
进一步地,所述三级氯化釜、结晶釜和氟化釜均设置有尾气吸收装置。
进一步地,所述第二精馏装置设置有反应副产品收集装置。
一种2-氯-3-三氟甲基吡啶的制备方法,包括以下步骤:
(1)将2-氯-3-甲基吡啶溶解于有机溶剂中,随后通入氯气进行反应,得到氯化粗产品,将所述氯化粗产品进行减压精馏、结晶,分离得到2-氯-3-三氯甲基吡啶;
(2)向所述2-氯-3-三氯甲基吡啶中通入氟化氢进行反应,得到反应产物,向所述反应产物中加入水和碱进行反应,静置分层,得到氟化粗产品;
(3)将所述氟化粗产物进行分离提纯处理,得到分离提纯后产物,将所述分离提纯后产物进行减压精馏,得到所述2-氯-3-三氟甲基吡啶。
进一步的,步骤(1)中,所述有机溶剂为氯苯、四氯化碳、二氯苯或邻二氯苯中的任意一种。
进一步的,步骤(1)中,所述2-氯-3-三氟甲基吡啶与所述有机溶剂的摩尔比为1:(2-10)。
进一步的,步骤(1)中,所述通入氯气的速度为38-42kg/h,所述氯化反应为多级氯化过程;所述反应的温度为100-160℃,所述反应时间为10-12h,所述反应压力为常压。
进一步的,所述多级氯化过程为二级氯化过程、三级氯化过程或四级氯化过程中的任意一种。
进一步的,所述三级氯化过程为:将溶于有机溶剂中的2-氯-3-甲基吡啶分三等份,分别投入三个反应釜,氯气依次通过三个反应釜,反应结束后,从一级反应釜取出氯化粗产品进行精馏处理;将二级反应釜中氯化粗产品导入一级反应釜中,将三级反应釜中氯化粗产品导入二级反应釜中,再次向三级反应釜中投入新的溶于有机溶剂中的2-氯-3-甲基吡啶,继续通入氯气继续反应;通过三级氯化过程,能够有效节约氯气使用量,且同时可以使反应更加充分完全,提高反应产物的收率。
进一步的,步骤(1)中,所述减压精馏的压力为-0.08mpa至-0.02mpa,所述减压精馏中140-160℃的馏分即为所述2-氯-3-三氯甲基吡啶。
进一步的,步骤(2)中,当通入氟化氢时,所述2-氯-3-三氯甲基吡啶的温度为78-82℃。
进一步的,步骤(2)中,所述2-氯-3-三氯甲基吡啶与所述氟化氢的摩尔比为1:(7-20)。
进一步的,步骤(2)中,所述通入氟化氢进行反应的温度为160-210℃,所述反应时间为8-12h,所述反应压力为3-9mpa。
进一步的,步骤(2)中,所述水的加入量为所述反应产物质量的90%-110%;所述加碱将体系ph调整至6.9-7.1,所述碱为氢氧化钠或氢氧化钾中的任意一种。加入水和碱的主要目的为去除反应体系中的氟化氢和氯化氢。
进一步的,步骤(3)中,所述分离提纯处理方法为离心、结晶或压滤中的任意一种;所述减压精馏的循环水温度为35-40℃,所述减压精馏的压力为-0.99mpa至-0.08mpa,所述减压精馏中100-150℃的馏分即为所述2-氯-3-三氟甲基吡啶。
本实用新型的上述技术方案相比现有技术具有以下优点:
本实用新型所述2-氯-3-三氟甲基吡啶的合成装置通过依次连通的多级氯化釜的设置,能够最高效利用氯气,避免了氯气的浪费,且同时可以使反应更加充分完全,提高反应产物的收率;通过第一精馏装置,能够大幅度提高中间反应产物2-氯-3-三氯甲基吡啶的纯度,进而可以有效进行氟化反应,间接提高产品收率;通过结晶釜能够进一步提高中间产物2-氯-3-三氯甲基吡啶的纯度;通过所述氟化釜能够高效氟化2-氯-3-三氯甲基吡啶,充分利用氟化氢,进一步降低合成成本;最后通过中和釜和第二精馏装置能够高效纯化终产物2-氯-3-三氟甲基吡啶,最终安全高效的得到高纯度产品。
本实用新型所述合成装置能够有效保证反应的顺利进行,并且有效提升了合成过程的安全性。经过本实用新型所述的合成设备制备得到的2-氯-3-三氟甲基吡啶纯度为99%以上,收率为80%以上。
本实用新型所述合成设备能够实现不用催化剂即可制备高纯度2-氯-3-三氟甲基吡啶产品的需求,降低合成成本的同时还大幅度提升生产安全;利用本实用新型所述合成设备的合成过程简单高效,原料成本低,所得产品纯度高,保证生产安全性,易于工业化推广。
附图说明
为了更清楚地说明本实用新型实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本实用新型的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是本实用新型实施例1所述2-氯-3-三氟甲基吡啶合成装置的结构示意图;
图中1-三级氯化釜;110-第一进料口;120-第一出料口;2-二级氯化釜;21-第二进料口;22-第二出料口;23-第一倒料口;3-一级氯化釜;31-第三进料口;32-第三出料口;33-第二倒料口;34-氯气导入口;4-第一精馏装置;41-第四进料口;42-第四出料口;5-结晶釜;51-第五进料口;52-第五出料口;6-氟化釜;61-氟化氢导入口;62-第六出料口;63-第六进料口;7-中和釜;71-水导入口;72-第七出料口;73-第七进料口;74-碱导入口;8-第二精馏装置;81-第八进料口;82-成品出料口;9-第一氯气通道;10-第二氯气通道;11-搅拌装置;12-回流管道;
图2是本实用新型实施例1所述三级氯化的装置图。
具体实施方式
为使本实用新型的目的、技术方案和优点更加清楚,下面将对本实用新型的技术方案进行详细的描述。显然,所描述的实施例仅仅是本实用新型一部分实施例,而不是全部的实施例。基于本实用新型中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本实用新型所保护的范围。
实施例1
本实施例提供一种2-氯-3-三氟甲基吡啶的合成装置,如图1和图2所示,包括依次连通设置的三级氯化釜1、二级氯化釜2、一级氯化釜3、第一精馏装置4、结晶釜5、氟化釜6、中和釜7、第二精馏装置8;所述三级氯化釜1上端设置有第一进料口110,下端设置有第一出料口120;所述二级氯化釜2上端设置有第二进料口21和第一倒料口23,下端设置有第二出料口22;所述一级氯化釜3上端设置有第三进料口31、第二倒料口33和氯气导入口34,下端设置有第三出料口32;所述三级氯化釜1和二级氯化釜2之间设置有第一氯气通道9,所述二级氯化釜2和一级氯化釜3之间设置有第二氯气通道10;
所述第一精馏装置4设置有第四进料口41和第四出料口42,所述第四进料口41与第三出料口32连通设置;所述结晶釜5设置有第五进料口51和第五出料口52,所述第五进料口51与第四出料口42连通设置;所述氟化釜设置有氟化氢导入口61、第六出料口62和第六进料口63,所述第六进料口63与第五出料口52连通设置;所述中和釜7设置有水导入口71、第七出料口72、第七进料口73和碱导入口74,所述第七进料口73与第六出料口62连通设置;所述第二精馏装置8设置有第八进料口81和成品出料口82,所述第八进料口81与第七出料口72连通设置。
本实施例提供一种2-氯-3-三氟甲基吡啶的制备方法,具体处理方法包括以下步骤:
(1)将1000g纯度为99%的2-氯-3-甲基吡啶用1731g氯苯溶解后平均分为三份,分别于第一进料口110、第二进料口21和第三进料口31投入三级氯化釜1、二级氯化釜2和一级氯化釜3中,调整反应温度至100℃,从氯气导入口34中以38-42kg/h的速度向一级反应釜3通入氯气,氯气经过第一氯气通道9和第二氯气通道10分别进入二级反应釜2和三级反应釜1,持续通入氯气保持10小时,进行氯化反应,得到氯化粗产品;将所述一级反应釜3中的氯化粗产品经第三出料口32和第四进料口41投入第一精馏装置4中进行减压精馏,将二级反应釜2中所得氯化粗产品导入一级反应釜3中,将三级反应釜1中所得氯化粗产品导入二级反应釜2中,再次向三级反应釜1中投入新的溶于有机溶剂中的2-氯-3-甲基吡啶,继续通入氯气继续反应;第一精馏装置4中的减压精馏控制压力为-0.02mpa,收集150℃馏分,经由第四出料口42和第五进料口51进入结晶釜5中进行结晶,收集结晶,得到1580.6g2-氯-3-三氯甲基吡啶,收率88.7%,含量94.9%,减压精馏后的反应液经由回流管道12回流至一级反应釜中继续反应。
(2)保持氟化釜6的温度在80℃,将所述2-氯-3-三氯甲基吡啶经第五出料口52和第六进料口63投入到氟化釜6中,投料完毕后,将氟化釜6温度提高至160℃,保持压力为3mpa,从氟化氢导入口61通入981g氟化氢,反应8小时,得到反应产物;反应完毕降温后缓慢排压3个小时,用氮气赶出酸气,后用氮气压料,将反应压力降低至常压;将所述反应产物经第六出料口62和第七进料口73投入到中和釜7中,向所述反应产物中通过水导入口71加入为所述反应产物质量90%的水,并通过碱导入口74加入氢氧化钠将反应体系ph调整至6.9,去除氟化氢和氯化氢,静置分层后得到氟化粗产品。
(3)将所述氟化粗产品通过第七出料口72和第八进料口81投入到第二精馏装置8中,首先所述氟化粗产品进行压滤处理,得到分离提纯后产物;随后将所述分离提纯后产物进行减压精馏,减压精馏的压力保持在-0.08mpa,冷凝器循环水水温为35℃;收集100℃馏分,最终从成品出料口82中收集到824.2g2-氯-3-三氟甲基吡啶,经过气相色谱分析,收率为81.3%,含量为99%。
实施例2
本实施例提供一种2-氯-3-三氟甲基吡啶的合成装置,包括依次连通设置的三级氯化釜1、二级氯化釜2、一级氯化釜3、第一精馏装置4、结晶釜5、氟化釜6、中和釜7、第二精馏装置8;所述三级氯化釜1上端设置有第一进料口110,下端设置有第一出料口120;所述二级氯化釜2上端设置有第二进料口21和第一倒料口23,下端设置有第二出料口22;所述一级氯化釜3上端设置有第三进料口31、第二倒料口33和氯气导入口34,下端设置有第三出料口32;所述三级氯化釜1和二级氯化釜2之间设置有第一氯气通道9,所述二级氯化釜2和一级氯化釜3之间设置有第二氯气通道10;
所述第一精馏装置4设置有第四进料口41和第四出料口42,所述第四进料口41与第三出料口32连通设置;所述结晶釜5设置有第五进料口51和第五出料口52,所述第五进料口51与第四出料口42连通设置;所述氟化釜设置有氟化氢导入口61、第六出料口62和第六进料口63,所述第六进料口63与第五出料口52连通设置;所述中和釜7设置有水导入口71、第七出料口72、第七进料口73和碱导入口74,所述第七进料口73与第六出料口62连通设置;所述第二精馏装置8设置有第八进料口81和成品出料口82,所述第八进料口81与第七出料口72连通设置;所述三级氯化釜1、二级氯化釜2、一级氯化釜3、结晶釜5、氟化釜6和中和釜7中均含有搅拌装置11;所述一级氯化釜3和第一精馏装置4之间设置有回流管道12;所述三级氯化釜1、结晶釜5和氟化釜6均设置有尾气吸收装置;所述第二精馏装置8设置有反应副产品收集装置。
本实施例提供一种2-氯-3-三氟甲基吡啶的制备方法,具体处理方法包括以下步骤:
(1)将1000g纯度为99%的2-氯-3-甲基吡啶用3463g二氯苯溶解后平均分为三份,分别于第一进料口110、第二进料口21和第三进料口31投入三级氯化釜1、二级氯化釜2和一级氯化釜3中,调整反应温度至115℃,从氯气导入口34中以42kg/h的速度向一级反应釜3通入氯气,氯气经过第一氯气通道9和第二氯气通道10分别进入二级反应釜2和三级反应釜1,持续通入氯气保持10.5小时,进行氯化反应,得到氯化粗产品;将所述一级反应釜3中的氯化粗产品经第三出料口32和第四进料口41投入第一精馏装置4中进行减压精馏,将二级反应釜2中所得氯化粗产品导入一级反应釜3中,将三级反应釜1中所得氯化粗产品导入二级反应釜2中,再次向三级反应釜1中投入新的溶于有机溶剂中的2-氯-3-甲基吡啶,继续通入氯气继续反应;第一精馏装置4中的减压精馏控制压力为-0.03mpa,收集150℃馏分,经由第四出料口42和第五进料口51进入结晶釜5中进行结晶,收集结晶,得到1610.9g2-氯-3-三氯甲基吡啶,收率90.4%,含量95.1%,减压精馏后的反应液经由回流管道12回流至一级反应釜中继续反应。
(2)保持氟化釜6的温度在85℃,将所述2-氯-3-三氯甲基吡啶经第五出料口52和第六进料口63投入到氟化釜6中,投料完毕后,将氟化釜6温度提高至170℃,保持压力为3.5mpa,从氟化氢导入口61通入1428g氟化氢,反应9小时,得到反应产物;反应完毕降温后缓慢排压4个小时,用氮气赶出酸气,后用氮气压料,将反应压力降低至常压;将所述反应产物经第六出料口62和第七进料口73投入到中和釜7中,向所述反应产物中通过水导入口71加入为所述反应产物质量90%的水,并通过碱导入口74加入氢氧化钠将反应体系ph调整至6.9,去除氟化氢和氯化氢,静置分层后得到氟化粗产品。
(3)将所述氟化粗产品通过第七出料口72和第八进料口81投入到第二精馏装置8中,首先所述氟化粗产品进行结晶处理,得到分离提纯后产物;随后将所述分离提纯后产物进行减压精馏,减压精馏的压力保持在-0.20mpa,冷凝器开循环水水温为35℃。收集100℃馏分,最终从成品出料口82中收集到844.4g2-氯-3-三氟甲基吡啶,经过气相色谱分析,收率为83.3%,含量为99.2%。
实施例3
本实施例提供一种2-氯-3-三氟甲基吡啶的合成装置,包括依次连通设置的三级氯化釜1、二级氯化釜2、一级氯化釜3、第一精馏装置4、结晶釜5、氟化釜6、中和釜7、第二精馏装置8;所述三级氯化釜1上端设置有第一进料口110,下端设置有第一出料口120;所述二级氯化釜2上端设置有第二进料口21和第一倒料口23,下端设置有第二出料口22;所述一级氯化釜3上端设置有第三进料口31、第二倒料口33和氯气导入口34,下端设置有第三出料口32;所述三级氯化釜1和二级氯化釜2之间设置有第一氯气通道9,所述二级氯化釜2和一级氯化釜3之间设置有第二氯气通道10;
所述第一精馏装置4设置有第四进料口41和第四出料口42,所述第四进料口41与第三出料口32连通设置;所述结晶釜5设置有第五进料口51和第五出料口52,所述第五进料口51与第四出料口42连通设置;所述氟化釜设置有氟化氢导入口61、第六出料口62和第六进料口63,所述第六进料口63与第五出料口52连通设置;所述中和釜7设置有水导入口71、第七出料口72、第七进料口73和碱导入口74,所述第七进料口73与第六出料口62连通设置;所述第二精馏装置8设置有第八进料口81和成品出料口82,所述第八进料口81与第七出料口72连通设置;所述三级氯化釜1、二级氯化釜2、一级氯化釜3、结晶釜5、氟化釜6和中和釜7中均含有搅拌装置11;所述一级氯化釜3和第一精馏装置4之间设置有回流管道12;所述三级氯化釜1、结晶釜5和氟化釜6均设置有尾气吸收装置;所述第二精馏装置8设置有反应副产品收集装置。
本实施例提供一种2-氯-3-三氟甲基吡啶的制备方法,具体处理方法包括以下步骤:
(1)将1000g纯度为99%的2-氯-3-甲基吡啶用5194g四氯化碳溶解后平均分为三份,分别于第一进料口110、第二进料口21和第三进料口31投入三级氯化釜1、二级氯化釜2和一级氯化釜3中,调整反应温度至130℃,从氯气导入口34中以39kg/h的速度向一级反应釜3通入氯气,氯气经过第一氯气通道9和第二氯气通道10分别进入二级反应釜2和三级反应釜1,持续通入氯气保持11小时,进行氯化反应,得到氯化粗产品;将所述一级反应釜3中的氯化粗产品经第三出料口32和第四进料口41投入第一精馏装置4中进行减压精馏,将二级反应釜2中所得氯化粗产品导入一级反应釜3中,将三级反应釜1中所得氯化粗产品导入二级反应釜2中,再次向三级反应釜1中投入新的溶于有机溶剂中的2-氯-3-甲基吡啶,继续通入氯气继续反应;第一精馏装置4中的减压精馏控制压力为-0.05mpa,收集150℃馏分,经由第四出料口42和第五进料口51进入结晶釜5中进行结晶,收集结晶,得到1621.6g2-氯-3-三氯甲基吡啶,收率91.0%,含量96.1%,减压精馏后的反应液经由回流管道12回流至一级反应釜中继续反应。
(2)保持氟化釜6的温度90℃,将所述2-氯-3-三氯甲基吡啶经第五出料口52和第六进料口63投入到氟化釜6。投料完毕后,将氟化釜6温度提高至185℃,保持压力为4mpa,从氟化氢导入口61通入2012g氟化氢,反应10小时,得到反应产物。反应完毕降温后缓慢排压6.5个小时,用氮气赶出酸气,后用氮气压料,将反应压力降低至常压;将所述反应产物经第六出料口62和第七进料口73投入到中和釜7中,向所述反应产物中通过水导入口71加入为所述反应产物质量110%的水,并通过碱导入口74加入氢氧化钠将反应体系ph调整至7.1,去除氟化氢和氯化氢,静置分层后得到氟化粗产品。
(3)将所述氟化粗产品通过第七出料口72和第八进料口81投入到第二精馏装置8中,首先所述氟化粗产品进行压滤处理,得到分离提纯后产物;随后将所述分离提纯后产物进行减压精馏,减压精馏的压力保持在-0.61mpa,冷凝器开循环水水温为35℃。收集100℃馏分,最终从成品出料口82中收集到862.7g2-氯-3,3-氟甲基吡啶,经过气相色谱分析,收率为85.1%,含量为99.5%。
实施例4
本实施例提供一种2-氯-3-三氟甲基吡啶的合成装置,包括依次连通设置的三级氯化釜1、二级氯化釜2、一级氯化釜3、第一精馏装置4、结晶釜5、氟化釜6、中和釜7、第二精馏装置8;所述三级氯化釜1上端设置有第一进料口110,下端设置有第一出料口120;所述二级氯化釜2上端设置有第二进料口21和第一倒料口23,下端设置有第二出料口22;所述一级氯化釜3上端设置有第三进料口31、第二倒料口33和氯气导入口34,下端设置有第三出料口32;所述三级氯化釜1和二级氯化釜2之间设置有第一氯气通道9,所述二级氯化釜2和一级氯化釜3之间设置有第二氯气通道10;
所述第一精馏装置4设置有第四进料口41和第四出料口42,所述第四进料口41与第三出料口32连通设置;所述结晶釜5设置有第五进料口51和第五出料口52,所述第五进料口51与第四出料口42连通设置;所述氟化釜设置有氟化氢导入口61、第六出料口62和第六进料口63,所述第六进料口63与第五出料口52连通设置;所述中和釜7设置有水导入口71、第七出料口72、第七进料口73和碱导入口74,所述第七进料口73与第六出料口62连通设置;所述第二精馏装置8设置有第八进料口81和成品出料口82,所述第八进料口81与第七出料口72连通设置;所述三级氯化釜1、二级氯化釜2、一级氯化釜3、结晶釜5、氟化釜6和中和釜7中均含有搅拌装置11;所述一级氯化釜3和第一精馏装置4之间设置有回流管道12;所述三级氯化釜1、结晶釜5和氟化釜6均设置有尾气吸收装置;所述第二精馏装置8设置有反应副产品收集装置。
本实施例提供一种2-氯-3-三氟甲基吡啶的制备方法,具体处理方法包括以下步骤:
(1)将1000g纯度为99%的2-氯-3-甲基吡啶用2126g邻二氯苯溶解后平均分为三份,分别于第一进料口110、第二进料口21和第三进料口31投入三级氯化釜1、二级氯化釜2和一级氯化釜3中,调整反应温度至130℃,从氯气导入口34中以40kg/h的速度向一级反应釜3通入氯气,氯气经过第一氯气通道9和第二氯气通道10分别进入二级反应釜2和三级反应釜1,持续通入氯气保持11小时,进行氯化反应,得到氯化粗产品;将所述一级反应釜3中的氯化粗产品经第三出料口32和第四进料口41投入第一精馏装置4中进行减压精馏,将二级反应釜2中所得氯化粗产品导入一级反应釜3中,将三级反应釜1中所得氯化粗产品导入二级反应釜2中,再次向三级反应釜1中投入新的溶于有机溶剂中的2-氯-3-甲基吡啶,继续通入氯气继续反应;第一精馏装置4中的减压精馏控制压力为-0.07mpa,收集150℃馏分,经由第四出料口42和第五进料口51进入结晶釜5中进行结晶,收集结晶,得到1621.6g2-氯-3-三氯甲基吡啶,收率91.0%,含量96.1%,减压精馏后的反应液经由回流管道12回流至一级反应釜中继续反应。
(2)保持氟化釜6的温度95℃,将所述2-氯-3-三氯甲基吡啶经第五出料口52和第六进料口63投入到氟化釜6中,投料完毕后,将氟化釜6温度提高至195℃,保持压力为4.5mpa,从氟化氢导入口61通入2443g氟化氢,反应11小时,得到反应产物;反应完毕降温后缓慢排压8个小时,用氮气赶出酸气,后用氮气压料,将反应压力降低至常压;将所述反应产物经第六出料口62和第七进料口73投入到中和釜7中,向所述反应产物中通过水导入口71加入同等质量的水,并通过碱导入口74加入氢氧化钠将反应体系ph调整至7,去除氟化氢和氯化氢,静置分层后得到氟化粗产品。
(3)将所述氟化粗产品通过第七出料口72和第八进料口81投入到第二精馏装置8中,首先所述氟化粗产品进行离心处理,得到分离提纯后产物;随后将所述分离提纯后产物进行减压精馏,真空度需要保持在-0.72mpa,冷凝器开循环水水温为35℃。收集100℃馏分,最终从成品出料口82中收集到856.6g2-氯-3-三氟甲基吡啶,经过气相色谱分析,收率为84.5%,含量为99.4%。
实施例5
本实施例提供一种2-氯-3-三氟甲基吡啶的合成装置,包括依次连通设置的三级氯化釜1、二级氯化釜2、一级氯化釜3、第一精馏装置4、结晶釜5、氟化釜6、中和釜7、第二精馏装置8;所述三级氯化釜1上端设置有第一进料口110,下端设置有第一出料口120;所述二级氯化釜2上端设置有第二进料口21和第一倒料口23,下端设置有第二出料口22;所述一级氯化釜3上端设置有第三进料口31、第二倒料口33和氯气导入口34,下端设置有第三出料口32;所述三级氯化釜1和二级氯化釜2之间设置有第一氯气通道9,所述二级氯化釜2和一级氯化釜3之间设置有第二氯气通道10;
所述第一精馏装置4设置有第四进料口41和第四出料口42,所述第四进料口41与第三出料口32连通设置;所述结晶釜5设置有第五进料口51和第五出料口52,所述第五进料口51与第四出料口42连通设置;所述氟化釜设置有氟化氢导入口61、第六出料口62和第六进料口63,所述第六进料口63与第五出料口52连通设置;所述中和釜7设置有水导入口71、第七出料口72、第七进料口73和碱导入口74,所述第七进料口73与第六出料口62连通设置;所述第二精馏装置8设置有第八进料口81和成品出料口82,所述第八进料口81与第七出料口72连通设置;所述三级氯化釜1、二级氯化釜2、一级氯化釜3、结晶釜5、氟化釜6和中和釜7中均含有搅拌装置11;所述一级氯化釜3和第一精馏装置4之间设置有回流管道12;所述三级氯化釜1、结晶釜5和氟化釜6均设置有尾气吸收装置;所述第二精馏装置8设置有反应副产品收集装置。
本实施例提供一种2-氯-3-三氟甲基吡啶的制备方法,具体处理方法包括以下步骤:
(1)将1000g纯度为99%的2-氯-3-甲基吡啶用8657g氯苯溶解后平均分为三份,分别于第一进料口110、第二进料口21和第三进料口31投入三级氯化釜1、二级氯化釜2和一级氯化釜3中,分别从第一进料口110、第二进料口21和第三进料口31中均加入2885g氯苯,调整反应温度至130℃,从氯气导入口34中以41kg/h的速度向一级反应釜3通入氯气,氯气经过第一氯气通道9和第二氯气通道10分别进入二级反应釜2和三级反应釜1,持续通入氯气保持11小时,进行三级氯化反应,得到氯化粗产品;将所述一级反应釜3中的氯化粗产品经第三出料口32和第四进料口41投入第一精馏装置4中进行减压精馏,将二级反应釜2中所得氯化粗产品导入一级反应釜3中,将三级反应釜1中所得氯化粗产品导入二级反应釜2中,再次向三级反应釜1中投入新的溶于有机溶剂中的2-氯-3-甲基吡啶,继续通入氯气继续反应;第一精馏装置4中的减压精馏控制压力为-0.08mpa,收集150℃馏分,经由第四出料口42和第五进料口51进入结晶釜5中进行结晶,收集结晶,得到1594.9g2-氯-3-三氯甲基吡啶,收率89.5%,含量94.5%,含量94.9%,减压精馏后的反应液经由回流管道12回流至一级反应釜中继续反应。
(2)保持氟化釜6的温度95℃,将所述2-氯-3-三氯甲基吡啶经第五出料口52和第六进料口63投入到氟化釜6中,投料完毕后,将氟化釜6温度提高至195℃,保持压力5mpa,从氟化氢导入口61通入2827g氟化氢,反应12小时,得到反应产物;反应完毕降温后缓慢排压10个小时,用氮气赶出酸气,后用氮气压料,将反应压力降低至常压;将所述反应产物经第六出料口62和第七进料口73投入到中和釜7中,向所述反应产物中通过水导入口71加入为所述反应产物质量105%的水,并通过碱导入口74加入氢氧化钠将反应体系ph调整至7,去除氟化氢和氯化氢,静置分层后得到氟化粗产品。
(3)将所述氟化粗产品通过第七出料口72和第八进料口81投入到第二精馏装置8中,首先所述氟化粗产品进行结晶处理,得到分离提纯后产物;随后将所述分离提纯后产物进行减压精馏,减压精馏的压力保持在-0.99mpa,冷凝器开循环水水温为35度。收集100℃馏分,最终从成品出料口82中收集到814.0g2-氯-3-三氟甲基吡啶,经过气相色谱分析,收率为80.3%,含量为99.1%。
以上所述,仅为本实用新型的具体实施方式,但本实用新型的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本实用新型揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本实用新型的保护范围之内。因此,本实用新型的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!