非水性分散剂的制作方法
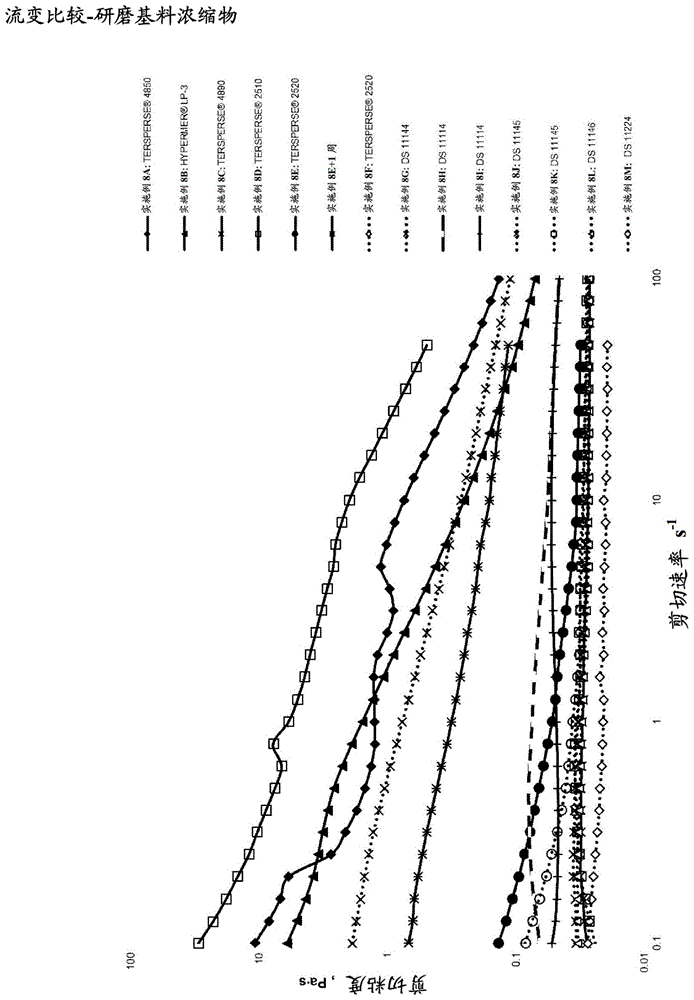
发明领域本发明通常涉及高度分散的农药在液体、尤其是非水性液体中的分散剂,其中稳定和有效的分散对于所形成的配制剂的最优性能和稳定性来说是关键的。所感兴趣的主要配制剂是非水性农药组合物,例如被croplifeinternational称作油可混溶的可流动浓缩物(“of”)的那些,和更特别地是油分散剂(“od”)。下文中将方便地描述关于固体的高度分散的农药在非水性液体中的分散体以及关于在其中使用聚羟基硬脂酸(“phsa”)的缩合衍生物的本发明,其显示出分散稳定性方面的改进。然而,本发明不被认为限于phsa衍生物。发明背景寻求制备高度分散的农药在非水性液体中的稳定分散体的配方研制者面临着诸多挑战。这些挑战往往是令人烦恼的储存期限稳定性行为的基础,所述储存期限稳定性行为往往表征了农用化学od配制剂。主要的挑战之一是将活性成分有效分散在非水性介质、特别是油基介质中的能力,它的成功通过帮助防止在储存期内的沉降和脱水收缩而对于od稳定性是至关重要的,和最终由此使得终端用户容易将该配制剂从容器转移到施用容器中。固体的高度分散的农药在非水性液体中的稳定的分散体典型地通过使用具体的非水性分散剂(“nad”)来促进,该非水性分散剂(“nad”)典型地含有一种或多种油溶性、亲脂性或亲油性结构部分,和合适的锚定结构部分。亲脂性结构部分倾向于可溶于它将典型地扩展到其中的外相中,这形成了有效空间阻挡,而锚定结构部分应当强力吸附到分散相上。通过使用特定分散剂来有效稳定非水性液体中的农药悬浮液的关键要素可论证地是分散剂的锚定结构部分与分散相的亲和性或相容性。并不令人惊讶地是缺乏协同作用可最终导致缺乏到期望的底物的足够的吸附性,分散剂从底物逐渐解吸和/或分散剂在外相中的过度溶解。如果发生任何这些行为,这将倾向于在完成配制的实施例中产生絮凝、沉降、相分离、剪切稠化或胀塑和/或络合物缔合稠化现象。然而,当处理农药在非水性液体中的分散时,分散剂性能的这个方面必须与大多数农药是有机的而非无机的概念以及与典型地从低到相当高的极性方面的显著变化并行考虑。归因于往往由高度分散的农药的界面提供的低能量表面,在非水性体系中分散剂的锚定结构部分会难以有效缔合和产生最优分散效果。在第一种情况下,困难来自于典型的分散剂所采用的两亲性质,其中在非水性分散体的情况下,锚定结构部分往往将是亲水性的。可以容易地想到低到中等极性的有机底物将是热力学不相容的,其中该分散剂处于或是差的吸附或是解吸的风险中,和因此可以增加分散体或悬浮液的随后的去稳定的可能性。在第二种情况下,通过引入更合适的且潜在不太亲水性的结合官能团来改进分散剂的吸附潜力,随后两亲性质的降低可以经由通过外相的潜在改进的溶剂化而阻碍吸附的热力学分量。这些概念在农用化学od配制剂技术中得到进一步放大,这归因于配制剂被设计成在施用之前在水中稀释的要求。不像农用化学of型体系所用的纯粹的非水性体系,必须加入乳化剂来促进该要求。然而,这些极性的界面活性添加剂的存在提供另外的去稳定影响,其中nad吸附可以受到锚定结构部分的溶解的影响,或nad还可以可预见地被替换。这些事件最终为nad解吸提供另外的诱因,其中随后的农药od配制剂将不再是储存稳定的。一种潜在地克服这个问题的方法是采用增效剂或分散增效剂。这些材料目的是吸附在分散相上和提供更合适的桥连界面,以提供与nad的锚定结构部分更好的相容性。然而,从开发和商业观点二者来看,使用增效剂增加了不想要的复杂性。可替代地,用于克服这种挑战的另一方法是使用高性能聚合物nad,其中锚定结构部分对于待分散的农用化学底物具有更大的特异性,理想地是通过引入共同的官能团,或引入可以在宽范围底物内提供吸附效用的特征。具有优异效用的高性能聚合物nad特别是梳形的那些:这些材料含有合适的锚定主链,在其上共价连接多个稳定的侧链。农用化学配方研制者确实可以利用先进的聚合物nad技术。合适的技术的巅峰实例是梳形聚合物nad4890,其是从huntsmancorporation可获得的,或13940,其是从thelubrizolcorporation可获得的。然而,在这些材料的锚定构造中所用的化学虽然提供了鲁棒且通用的吸附行为,但是在农用化学法规考虑方面是不受欢迎的。特别要注意的是低风险聚合物定义,例如由美国环保局标题第40号联邦法规(40codeoffederalregulations)(40cfr),具体是40cfr§723.250所提供的那些。因此需要可替代的策略来制备类似的高性能nad,该nad呈现出类似性能,但是其仍然落入任何商业上不利的法规限制的限度内并且考虑到工业优势或制造简明性。照此,本发明人已经检查了相对疏水性的锚定构造的效用,其从根本上补充了低到中等极性有机农药底物,但是其使用不是立即直观的,这归因于对于分散剂两亲性质的影响和随后关于吸附潜力的效果。因此,为了改进承诺借助于od配制剂的非水性外相提供更有效的农药的递送载体(其产生了在罐内的辅助性)的od配制剂技术的可利用性,本发明人已经发现理论上不太两亲性的,或相对更疏水性的非水性分散剂显示出令人惊讶的鲁棒分散性能。这已经通过在研磨基料浓缩物流变行为中的明显改进得以显示,其被表明由分散剂锚和分散相之间改进的亲和性产生。与现有技术相比,预期这种改进有助于促进更大的od配制剂稳定性,同时这些材料与往往用于稳定非水性分散体的其他分散剂相比也更容易处置和保持优异的溶解性。由于现有有限组的可商购农药的效力因诸如耐药性的问题而下降,以及由于更新更复杂的技术遭受日益降低的生物可利用性,诸如此类的改进的递送主题越来越重要。本发明寻求改善现有技术中遇到的问题中的至少之一。发明概述根据本发明的一方面,提供一种非水性分散剂(“nad”),其包含通过以有效反应化学计量将疏水性稳定体(“hse”)的加成物接枝到共聚物上而形成的缩合物,其中该共聚物包含具有芳族官能团的锚定结构部分。在本说明书中,本领域技术人员将理解下文中使用的术语“缩合物”表示在任何包括加成反应、缩合反应或取代反应的反应之后所获得的产物。术语“疏水性稳定体”将被本领域技术人员理解为表示具有合适分子量(其将典型地具有1000da或更大的分子量)的合适的疏水性、或可替代的溶剂相容性或油相容性的聚合物、低聚物或其他材料,其可以通过空间稳定化机理自身或当通过反应、缩合或否则用合适的试剂改性时,提供或促进非水性分散体特性。术语“共聚物”被理解为表示通过两种或更多种单体物类反应而制造的聚合物。术语“大分子单体”被理解为表示这样的大分子,其具有至少一个使得它能够充当单体的端基如烯属双键,因此使得该大分子能够参与聚合反应。如本文所用,在聚合物分散剂(无论水性还是非水性的)的上下文中,大分子典型地是通过空间稳定化机理而促进分散特性的稳定体,即疏水性稳定体,例如聚羟基硬脂酸(“phsa”)。术语“酸值”被理解为表示中和1g化学物质所需的以毫克计的氢氧化钾的质量。它应当通过精确称重待在锥形烧瓶中检查的样品的代表性质量来测定。溶解溶剂然后以1:1甲苯:乙醇(v/v)来制备,补足到100ml体积。向其中加入5滴的1%酚酞指示剂溶液,随后加入几滴0.1mkoh(在甲醇中),直到注意到粉红色的第一迹象。溶解溶剂然后与待检查的样品合并,并且当样品完全溶解时(其中可以需要热量以帮助溶解),将溶液用标准化的0.1m的koh甲醇溶液滴定。酸值使用以下等式确定:酸值=(t×m×56.1)/w,其中t是以ml计的滴定度;m是在甲醇溶液中标准化koh的摩尔浓度;和w是以克计的样品重量。因此,酸值单位是mgkoh/g。如本领域技术人员所理解的,如果单个聚合物分子的酸官能团的数目是已知的或可以被定义,则术语“酸值”可以用于确定聚合物的引导分子量。即,对于phsa来说,通式是mw=a*56100/av,其中a=酸性基团的数目,56100单位是毫当量koh或mgkoh/mol,和av是mgkoh/g。通过简单的重排,可以获得以g/mol计的分子量近似值。hse优选选自聚羟基硬脂酸(“phsa”)、聚异丁烯琥珀酸酐(“pibsa”)、聚己内酯等,其中形成了具有特定分子量范围的加成物的衍生物。最优选地,hse是分子量大于1000da的phsa。在以有效反应化学计量与共聚物反应之前,hse如phsa优选首先通过与连接体底物反应来改性,该连接体底物选自二醇、烷醇胺、醚胺或亚乙基胺。以此方式,hse可以合适地制备以用于接枝到共聚物上。在另一优选的形式中,连接体底物选自聚氧化亚烷基,例如单乙二醇(“meg”)、二乙二醇(“deg”)、三乙二醇(“teg”)、聚乙二醇(“peg”)或其中式为ho[-ch2-ch-r-o-]n-h,r=h或ch3且n=1-100的聚氧化亚烷基;或甘醇胺(glycolamine)如二甘醇胺(“dga”)或三甘醇胺(“tga”)等;或聚醚胺,例如选自d系列的聚醚胺,即胺封端的聚氧化亚丙基,或ed系列,即主要具有聚乙二醇主链的二胺(该主链具有端胺),或4-羟基苯胺,或单乙醇胺(“mea”),或单异丙醇胺(“mipa”),乙二胺(“eda”),三亚乙基四胺(“teta”),或四亚乙基五胺(“tepa”)。最优选地,hse是phsa并且接枝促进的连接体底物是mea,其中hse加成物是phsa-单乙醇酰胺。类似地,使用包含聚异丁烯琥珀酸酐(“pibsa”)的加成物的衍生物,特别是pibsa与甘醇胺如dga或tga、单乙醇胺(“mea”)、乙二胺,或聚醚胺如选自d或ed系列的聚醚胺,或4-羟基苯胺进行反应的加成物的衍生物,也可以有利地提供相同的效果。在聚己内酯的情况下,加成物的衍生物可以典型地通过己内酯与合适的脂肪醇或脂肪胺反应来形成,这产生了具有大于1000da的合适的分子量的产物,其可以直接接枝到合适的共聚物上。在另一优选的形式中,hse的加成物如下来制备:由phsa与n-羟乙基丙烯酰胺(“heaa”)、n-羟乙基甲基丙烯酰胺(“hemam”)、丙烯酸2-羟乙酯(“hea”)、甲基丙烯酸2-羟乙酯(“hema”)、4-乙烯基苯胺、4-乙烯基苯酚、烯丙基醇如2-丙烯-1-醇等,或烯丙基胺如3-氨基-1-丙烯等在适当的条件下进行缩合反应,以形成产生的具有烯属不饱和末端的大分子单体,其可以与选自苯乙烯、马来酸酐、甲基丙烯酸或甲基丙烯酸甲酯的单体经由典型的自由基聚合技术来进一步共聚。虽然所形成的反应产物与优选的实施方案不是结构上相同的,但是预期显示出类似的应用性能。类似的应用效果还可以通过用简单脂肪醇如c6-c22饱和的以及单不饱和和多不饱和的脂肪醇;和脂肪胺如c12-c22饱和的以及单不饱和和多不饱和的脂肪胺代替期望的hse加成物来实现。然而,它们的较小的分子量将可能降低它们作为hse替代物的效力,这归因于不足的空间特性。优选地,在与单乙醇胺预缩合中所用的phsa将理想地具有大于1000da的分子量,其通过约25-50mgkoh/g的酸值来确定。在一种优选的形式中,其中共聚物的锚定结构部分的芳族官能团通过苯乙烯-马来酸酐(“sma”)共聚物来提供,优选的sma共聚物是-1000(polyscopepolymersbv),其具有1:1的苯乙烯:马来酸酐比。然而,共聚物也可以选自-2000(polyscopepolymersbv),其具有2:1的苯乙烯:马来酸酐比;-3000(polyscopepolymersbv),其具有3:1的苯乙烯:马来酸酐比;ef40、60和80(polyscopepolymersbv),其中sma共聚物与hse的加成物反应以形成化学计量比范围为1:1-8:1并且分子量(mn)范围为1000-20000da的缩合物。虽然使用sma共聚物是优选的,但是本发明还涉及使用具有芳族官能团的替代选项,包括苯乙烯、α-甲基-苯乙烯等的共聚物,其与丙烯酸、巴豆酸、衣康酸、甲基丙烯酸和甲基丙烯酸甲酯形成加成物。在一种最优选的形式中,本发明的缩合物具有酸性的和/或阴离子型性质。产生改进性能的最优选的nad是其中缩合物是酸性和阴离子型性质二者的,并且其中它包括具有芳族官能团的锚定结构部分的nad。这通过将phsa-单乙醇酰胺加成物的化学计量调节到小于基于共聚物的酸当量的化学计量当量来实现,其中如果需要的话,则残留酸度可以用合适的碱性/碱试剂进一步中和来形成期望的盐。在另一优选的形式中,通过调节反应化学计量,缩合物进一步包括磺酸、膦酸或磷酸官能团。在一种形式中,这是通过hse和至少一种带有磺酸盐、带有磷酸盐或带有膦酸盐接枝剂的加成物与共聚物的共缩合来实现。在一种更优选的形式中,phsa-单乙醇酰胺加成物的化学计量可以减少到小于基于共聚物的酸当量的化学计量当量,以允许带有磺酸、膦酸或磷酸的试剂的共缩合。带有磺酸的试剂优选选自牛磺酸、羟乙磺酸、4-氨基苯磺酸、4-羟基苯磺酸或1-羟基-2-丙烷磺酸或任何它们各自的盐。带有膦酸或磷酸的试剂优选选自氨基甲基膦酸、氨基乙基膦酸、磷酸乙醇胺或任何它们各自的盐。如果需要的话,则通过使用未中和的带有酸的试剂实现的缩合物可以稍后用合适的碱性/碱试剂中和以形成期望的盐。在另一优选的形式中,类似的官能化非接枝反应产物也可以经由phsa与heaa的缩合,而得自前述大分子单体hse的使用,其中烯属不饱和末端可以与适当量的选自苯乙烯、甲基丙烯酸、巴豆酸、衣康酸、马来酸酐、甲基丙烯酸、甲基丙烯酸甲酯或2-丙烯酰胺-2-甲基丙烷磺酸的单体经由典型的自由基聚合技术来共聚。在另一优选的形式中,对于优选的phsa-单乙醇酰胺来说,hse加成物可以直接接枝到现有的可商购共聚物上,该共聚物由但不限于苯乙烯或苯乙烯与马来酸酐等、或与2-丙烯酰胺-2-甲基丙烷磺酸等的加成物来制备。含有接枝到含芳族的主链锚上的残留量的磺酸结构部分的实例的开发已经表现出在表观底物亲和性方面甚至进一步的增加和因此特别是令人烦恼的分散相的改进的稳定化。这种将残留水平的阴离子型性质引入锚定结构部分中的方法是特别实际的,因为当用于脂族体系时,分散剂中多余的阴离子型性质的存在尽管潜在地在分散体稳定性方面是有益的,但是可以提供限制。这些限制主要涉及有限的溶解度、潜在的不期望的流变现象,和差的处置特质,其归因于富阴离子型nad的潜在的油脂状行为。本发明已经在特别是令人烦恼的有机农药化学的非水性分散体稳定性方面呈现出显著的改进。假定这可以在整体配制剂稳定性方面提供随后改进,整体配制剂稳定性已经成为阻止od技术的主流采用的工业上普遍的问题。本发明的主要优点涉及显著改进的高度分散的农药在非水性液体中的分散体的稳定性,其随后改进了所形成的od配制剂的稳定性。分散性能的改进通过非水性农药研磨基料浓缩物的视觉和流变观察来识别。通过使用由本公开内容所述的优选的反应产物而实现的关键的改进包括通过“降膜”主观tyndall散射评级和显微镜法所观察的可见絮凝的显著减少;浓缩物稳定性的改进,即缺乏异常的表现为糊或芥末状外观的去稳定作用;通过流变检查所观察的研磨基料浓缩物粘度的一致下降,其是改进的分散剂性能的指示。附图说明为了使本发明可以容易地理解并且付诸实践,现在将参考附图来提及本发明的实施方案。图仅是作为实例来提供。图1是显示如实施例8a至8m所示例的研磨基料浓缩物的流变比较图。图2是图1的展开或放大视图。图3描绘了初级研磨基料浓缩物系列a在20倍放大率下的显微照片图像。发明的详述在其最优选的形式中,缩合物是phsa-单乙醇酰胺加成物和苯乙烯-马来酸酐(“sma”)共聚物之间缩合的反应产物,其中优选的共聚物是sma-1000(polyscopepolymersbv),其具有1:1的苯乙烯:马来酸酐比并且mn=2100g/mol。phsa-单乙醇酰胺加成物的化学计量等价于,或最优选降低到小于基于共聚物酸值的化学计量当量。在与mea预缩合中使用的phsa将理想地具有大于1000g/mol的分子量,其通过约25-50mgkoh/g的酸值来确定。最优选地,将phsa-单乙醇酰胺加成物的化学计量降低到小于基于共聚物酸值的化学计量当量,以允许与牛磺酸共缩合。优选的实施方案反应物的优选结构如下:h-(oa)m-nh2(1)其中:a是乙烯或丙烯基团,和;m=≥1;其中:r1=c5-c30烷基或亚烷基;x=≥1;其中:k=1-8;m=1;和n是提供1000-20000的共聚物分子量mn的数字;和其中:x=n和n=2;或x=o和n=1;r1=c2-c30烷基或亚烷基,或取代或未取代的芳基。y=h或na。在一种优选的形式中,由反应物(1)和反应物(2)反应所产生的缩合物进一步与反应物(3)的共聚物分别以大约1:1化学计量比反应,以形成缩合物。更优选地,反应物(1)是单乙醇胺,反应物(2)是聚羟基硬脂酸和反应物(3)是sma-1000。在另一优选的形式中,由反应物(1)、反应物(2)反应所产生的缩合物和反应物(4)的ω-氨基或羟基-烷基磺酸与反应物(3)的共聚物分别以大约0.9:0.1:1化学计量比反应,以形成缩合物。更优选地,反应物(1)是单乙醇胺,反应物(2)是聚羟基硬脂酸,反应物(4)是2-氨基乙烷磺酸和反应物(3)是sma-1000。在又另一优选的形式中,上述缩合物用合适的试剂如naoh进一步中和。在又另一优选的形式中,由反应物(1)、反应物(2)反应所产生的缩合物和反应物(4)的ω-氨基或羟基-烷基磺酸的钠盐与反应物(3)的共聚物分别以大约0.9:0.1:1化学计量比反应,以形成缩合物。更优选地,反应物(1)是单乙醇胺,反应物(2)是聚羟基硬脂酸,反应物(4)是2-氨基乙烷磺酸钠盐和反应物(3)是sma-1000。实施例制备实施例1phsa-单乙醇酰胺加成物(“phsa-mea”)gphsa1502单乙醇胺58二甲苯158.8将在60℃烘箱中预热的phsa在氮气氛下装入2l带凸缘的烧瓶中。烧瓶作为回流过程,用装备有温度探针和恒温控制的加热罩的dean-stark分水器来设置。然后烧瓶装有mea和二甲苯,并且将反应混合物设定到170℃。当酸值平稳低于1mgkoh/g时,认为反应完成。随后,将dean-stark分水器的内容物排走和在没有任何溶剂除去的情形下分离产物。最终产物:深棕色粘性液体反应时间:440min估算的mw:1791.3g/mol酸值(起始):28.2mgkoh/g酸值(最终):0.72mgkoh/g固体:92.6%分离产率:1661.81g,96.6%实施例2以1:1化学计量比反应的sma-1000/phsa-单乙醇酰胺(“ds11144”)gphsa–mea衍生物(实施例1)424.10sma-100063.14二甲苯50.73钛酸四正丁酯(tnbt)0.517在氮气氛下向作为回流过程利用装备有温度探针、氮气入口、通用入/出端口和恒温控制的热油循环器的dean-stark分水器而设置的夹套式1lradley’sreactorreadylab反应器中装入如实施例1所述制备的熔融的phsa-mea加成物,并且将设定温度调节到50℃。当达到设定温度时,将固体sma-1000(polyscopepolymersbv)加入反应器中。当反应器中的混合物变成非均相时,将二甲苯加入dean-stark分水器和反应器中,并且将反应温度调节到150℃。在152℃下,反应混合物看起来已经溶解,并且将设定温度升高到190℃。将反应混合物在190℃下保持120分钟,并且要注意的是反应混合物变得更透明,颜色从哑光白色变成橙色。将反应混合物冷却到150℃,然后加入钛酸四正丁酯催化剂(“tnbt”、tnbt,dorfketalchemicals,llc),并且将设定温度调节到205℃。将反应混合物在这个温度下保持232分钟。在这段时间内,产物变得稍微更深。将产物冷却,然后在93℃下作为深棕色、橙色粘性液体从反应器烧瓶中分离。最终产物:深棕色、橙色粘性液体反应时间:628min酸值(起始):16.8mgkoh/g酸值(最终):4.7mgkoh/g固体:90.0%分离产率:479.8g,89%ftir/atr主峰:2923、2853、1731、1464、1174、700cm-1-brukeralpha粘度:不可测量实施例3以1:0.9:0.1化学计量比反应的sma-1000/phsa-单乙醇酰胺/牛磺酸(“ds11114”)在氮气氛下向作为回流过程用装备有温度探针、氮气入口、通用入/出端口和恒温控制加热罩的dean-stark分水器来设置的1l带凸缘的烧瓶中装入如实施例1所述制备的熔融的phsa-mea加成物,并且将温度设定到60℃。当达到设定温度时,dean-stark反应器装有在加热下溶解在水中的牛磺酸(scharlau,ar级)以及固体sma-1000并且用二甲苯洗涤。反应温度增加到150℃并且在该温度下保持90分钟,随后加入tnbt催化剂。然后将设定温度增加到205℃并且将反应混合物在这个温度下保持490分钟。在此时间段期间,在330分钟后加入第二量的tnbt催化剂。随后,使用氢氧化钠水溶液将磺酸结构部分部分地中和到50%摩尔当量的目标水平并且搅拌30分钟。将产物冷却,和然后在109℃下作为深棕色、橙色粘性液体从烧瓶中分离。最终产物:深棕色,橙色粘性液体酸值(起始):6.64mgkoh/g酸值(最终):0.95mgkoh/g固体:93.5%分离产率:408.47g,80%ftir/atr主峰:2923、2853、1731、1464、1174、700cm-1-brukeralpha粘度:2762cp@39℃–brookfield,#3转子,rpm=30实施例4以1:0.9:0.1化学计量比反应并且用naoh原位中和的sma-1000/phsa-单乙醇酰胺/牛磺酸(“ds11145”)在氮气氛下向作为回流过程用装备有温度探针、氮气入口、通用入/出端口和恒温控制加热罩的dean-stark分水器来设置的1l带凸缘的烧瓶中装入如实施例1所述制备的熔融的phsa-mea加成物,并且将温度设定到60℃。当达到设定温度时,向反应烧瓶中装入在加热下溶解在水中的牛磺酸以及固体sma-1000并且用二甲苯洗涤。将反应混合物的温度升高到124℃并且在回流下保持,直到它变成均相的。将反应温度增加到150℃和在该温度下保持150分钟,随后加入tnbt催化剂。将设定温度增加到205℃和将反应混合物在该温度下保持300分钟。加入第二量的催化剂和将反应在205℃设定点下持续583分钟,直到所测量的酸值平稳。随后,使用经由注射器直接加入反应器中的氢氧化钠水溶液将磺酸结构部分部分地中和到90%摩尔当量的目标水平并且在193℃下搅拌30分钟。然后将反应混合物放置在一边以冷却,其后将作为深橙色粘性液体的产物在108℃下从反应器烧瓶中分离。最终产物:深橙色粘性液体酸值(起始):6.60mgkoh/g酸值(最终):0.80mgkoh/g固体:87.7%分离产率:416.00g,89%ftir/atr主峰:2923,2853,1731,1464,1174,700cm-1-brukeralpha粘度:4610cp@38.5℃-brookfield,#3转子,rpm=20实施例5以1:0.9:0.1化学计量比反应的sma-1000/phsa-单乙醇酰胺/牛磺酸钠(“ds11145”)在氮气氛下向作为回流过程用装备有温度探针、氮气入口和通用入/出端口的dean-stark分水器来设置的1l带凸缘的烧瓶中装入熔融的phsa-mea衍生物,并且将温度设定到60℃。当达到设定温度时,向该反应器中装入牛磺酸钠以及固体sma-1000并且用exxsold60洗涤。设定温度设定到180℃和将反应混合物在该温度下保持大约9-10小时。然后将反应混合物放置在一边以冷却,其后将作为深橙色粘性液体的产物在大约100℃下从反应器烧瓶中分离。最终产物:棕色粘性液体酸值(最终):2.24mgkoh/g固体:90.7%分离产率:516.2g,92%实施例6以0.92:1.00:0.46化学计量比反应并且用naoh原位中和的sma-1000/phsa-单乙醇酰胺/牛磺酸(“ds11146”)gphsa–mea衍生物(实施例1)440.86sma-100027.78牛磺酸13.02水44.16二甲苯56.55钛酸四正丁酯1.07naoh,50%水溶液7.49使用作为回流过程用装备有温度探针、氮气入口、通用入/出端口和恒温控制加热罩的dean-stark分水器来设置的1l带凸缘的烧瓶。在氮气氛下烧瓶装有如实施例1所述制备的熔融phsa-mea加成物和将温度设定到60℃。反应器装有作为浆体溶解在热水中的牛磺酸,以及固体sma-1000并且用二甲苯洗涤。设定温度是150℃和保持150分钟。加入tnbt催化剂和将温度设定到205℃。加入第二量的催化剂和将反应在205℃的设定点下持续610分钟,其后所测量的酸值平稳。随后,使用经由注射器直接加入反应器中的氢氧化钠水溶液将磺酸结构部分部分地中和到90%摩尔当量的目标水平和在205℃下搅拌200分钟。然后将反应混合物放置在一边以冷却,其后将作为深棕色橙色粘性液体的产物在大约100℃下从反应器烧瓶中分离。最终产物:深棕色、橙色粘性液体酸值(起始):9.64mgkoh/g酸值(最终):7.40mgkoh/g固体:94.5%分离产率:384.25g,80%ftir/atr主峰:2923、2853、1731、1464、1174、700cm-1-brukeralpha粘度:7137cp@50℃-brookfield,#4转子,rpm=50实施例7以1:0.9:0.1化学计量比反应的sma-3000/phsa-单乙醇酰胺/牛磺酸钠(“ds11224”)gphsa–mea衍生物(实施例1)291.1sma-300035.3牛磺酸钠2.64exxsold6015.0在氮气氛下向作为回流过程用装备有温度探针、氮气入口和通用入/出端口的dean-stark分水器来设置的1l带凸缘的烧瓶中装入熔融的phsa-mea衍生物,并且将温度设定到60℃。当达到设定温度时,向反应器中加入牛磺酸钠以及固体sma-3000和用exxsold60洗涤。设定温度设定到180℃和将反应混合物在该温度下保持大约9-10小时。然后将反应混合物放置在一边以冷却,其后将作为深橙色粘性液体的产物在大约100℃下从反应器烧瓶中分离。最终产物:棕色粘性液体反应时间:780min。酸值(最终):3.40mgkoh/g固体:92.0%分离产率:317.1g,90%粘度:未测量实施例8施用实施例根据表1,在研磨基料浓缩物上进行评价以建立nad的基本性能。该研磨基料浓缩物通过如下来制备:将yubase3加入到适当预混的容器中,随后加入nad,然后加入活性成分。将混合物使用磁搅拌器短暂搅拌以形成粗浆体。然后将混合物送过laboratorymini100horizontalmill(engineeredmillsandmixers,inc.)。该过程包括将浓缩物缓慢进料到以500-1000rpm低转速运行的碾磨设备中,其中磨机研磨腔已经预先加载有60-80%总体积容量的1-1.6mm直径玻璃,或更优选的硅酸锆研磨介质,并且夹套的冷却剂温度是预设的,并且保持在15-25℃的外部控制的温度。在30-45分钟的时间段内将转速从2000缓慢增加到2500rpm,这产生了使用0至100研磨规,或8至0亥格曼规(hegmangauge)近似测定的平均(d0.5)粒度是大致5μm的浓缩物。粒度分析是另外通过显微镜近似测定的。浓缩物然后用于比较nad(包括现有的商业基准),其细节显示在下表2和3中。表1通用研磨基料浓缩物组成表2研磨基料浓缩物组成表3研磨基料浓缩物系列a的结果实施例研磨基料浓缩物的物理观察实施例8a中等粘性触变悬浮液,没有显著的随时间改变。实施例8b低到中等粘性触变悬浮液,没有显著的随时间改变。实施例8c低粘度悬浮液。没有显著的随时间改变。实施例8d初始低粘度,但是逐渐变成粘性发展到不可逆的硬质糊。实施例8e初始优异。但是缓慢发展到不可逆的粗砂质、糊状非均相混合物。实施例8f初始优异。但是缓慢发展到不可逆的粗砂质、糊状非均相混合物。实施例8g低粘度触变悬浮液,没有显著的随时间改变。实施例8h低粘度悬浮液。没有显著的随时间改变。实施例8i低粘度悬浮液。没有显著的随时间改变。实施例8j低粘度悬浮液。没有显著的随时间改变。实施例8k低粘度悬浮液。没有显著的随时间改变。实施例8l低粘度悬浮液。没有显著的随时间改变。实施例8m低粘度悬浮液。没有显著的随时间改变。结果讨论流变比较–研磨基料浓缩物非常像相关的水性农业悬浮液浓缩物(“sc”),od配制剂也可以经由制备研磨基料浓缩物来优先配制。对于是商业上可行的sc或od配制剂,它的研磨基料浓缩物需要具有合适的低粘度以允许有效粉碎,并且通常没有流变现象如过度的触变性或胀塑性,其倾向于导致生产低效或甚至失效。低研磨基料浓缩物粘度倾向于反映有效分散剂性能,其中所述性能当然不仅对于有效研磨是关键的,而且也是完成配制的浓缩悬浮液整体稳定化的一个关键支柱。然而,特别是对于农业od配制剂的情况来说,本发明人已经发现获得合适的低粘度研磨基料浓缩物,或借助于观察团聚和流变行为来确定的令人满意地稳定的研磨基料浓缩物通常是挑战性的。本发明人假定农用化学nad技术中潜在的缺陷以及它所产生的特定挑战是限制非水性od配制剂技术的增长的一个关键要素。方便地,在给定的油/溶剂混合物中根本nad性能的表征可以借助于研磨基料浓缩物的简单目视和流变评估来解决。这种方法的优点在于浓缩物将往往没有源自于其他关键配制剂要素如乳化和流变改性剂的存在的显著的复杂性。在这种情况下,在制备该研磨基料浓缩物的24小时内进行简单的目视评估,之后在随后几周内进行一般的再检查。流变实验经由malvernkinexusprorheometer使用40mm不锈钢平行板几何形状以150μm间隙在25℃的固定温度下进行。流变实验被限制到简单的流动曲线,其评价了剪切粘度比剪切速率。可以想到的是nad的有效性的改变将通过改变粒子-粒子动力学而影响分散体的流变特性。这些行为可以通过使用流变仪,通过流动类型或振荡测量来探测。在这种情况下,本发明人发现植物生长调节剂赛苯隆(thidiazuron)一致地显示出归因于有缺陷技术的差的分散剂性能的相同指示性特征,这已经在尝试开发含有其他农药活性成分的od配制剂时事先观察到。然而,赛苯隆用于评估潜在的通用nad性能的关键效用是这些特征伴随着容易观察到的流变异常,特别是不想要的触变性。这些改变容易通过更简单的流动评估来量化,其中随着低剪切粘度的数量级的增加,甚至nad性能的微小下降也容易观察到。图1和图2显示了改进的nad性能和显著降低的剪切粘度之间的相关性。立即显而易见的是使用现有的可利用的技术如2510、4850和lp-3产生了不合适的粘度,这可归因于絮凝,如图3所示。然而,其他可利用的技术如2520和4890确实设法提供了有效性能和触变性的显著下降。然而,2520如实施例8e所示随时间变劣,而4890未能满足低风险聚合物的标准,其限制了该技术和相关技术的商业可行性。实施例8g(ds11144)首先显示了令人惊讶的增强的nad性能,这可归因于通过使用sma主链的两亲性质的相对降低。ds11144,尽管基于相对锚定基团类似性的比较,预期概念上梳形接枝共聚物在功能上类似于更传统的a-b型分散剂4850,但是低剪切粘度降低到1/4至1/5。实施例8h至8m显示了通过借助于牛磺酸的共缩合来进一步衍生化而实现的明显的改进。使用ds11114、ds11145、ds11146和ds11124各自提供了有问题的触变性的有效去除和与基准4890相当的性能。更具体地,全部四种nad预期功能上类似于基准阴离子nad,lp-3,这同样基于锚定化学的简单比较,其中lp-3是明显更两亲性的,和其中离子性质表现为对整体性能非常有影响,这通过实施例8h和8i的流动曲线比较来证明。虽然相比较来说,ds11114、ds11145、ds11146和ds11224粘度下降到大约1/200,但是同样突出显示了通过利用sma主链而降低概念上两亲性质的令人惊讶的效果。此外,如实施例8m所示的ds11224的相对强的性能进一步突出显示了sma-锚令人惊讶的效力,其中借助于用sma-3000取代sma-1000,主链苯乙烯含量的3倍增加和随后两亲性质另外的下降预期产生显著的性能损失。显微镜法如上所述,流变现象直接与絮凝的存在相关,这归因于有缺陷的的分散剂性能。这种相关性借助于呈现了研磨基料浓缩物的显微照片图像的图3可目视观察。显微照相通过安装有附接的照相机系统的olympusbx43显微镜来实现。研磨基料浓缩物通过首先在大约10ml的yubase3中稀释3-4滴待观察的悬浮液来观察。然后将样品短暂搅拌,然后仔细逐滴加入到适当尺寸的玻片上,并且用盖玻片仔细盖上。为了支持流变性评估,分别使用ds11114、ds11145、ds11146和ds11224的实施例8h、8j、8l和8m的显微照片清楚地示出了材料性能的相对增加,这可归因于由于sma-基锚定结构部分令人惊讶的效力而消除的絮凝。在本说明书中使用术语“包含(包括)(comprise)”、“包含(包括)(comprises)”、“包含(包括)(comprising)”、“包括(include)”,“包括(includes)”、“包括(included)”或“包括(including)”的情况下,它们被解释为指定存在所提及的所述特征、整数、步骤或组分,但不排除存在或加入一种或多种其他特征、整数、步骤、组分或其组。此外,在说明书中提供的任何现有技术参考或声明不被用作承认这样的技术构成,或被理解为构成公知常识的一部分。本领域技术人员将理解除了具体示例的那些之外的材料和方法可以用于本发明的实践中,而无需借助于过度实验。任何这样的材料和方法的全部技术公知的功能等价物意图包括在本发明中。已经采用的术语和表达作为说明而非限制性术语来使用,并且在这样的术语和表达的使用中并非意图排除所示和所述的特征或其部分的任何等价物,但认识到各种改变在要求保护的发明的范围内是可能的。因此,应当理解虽然本发明已经通过实施例、优选的实施方案和任选的特征具体地公开,但是本领域技术人员可以借助于本文公开的构思的改动和变化,并且这样的改动和变化被认为在所附加权利要求所定义的本发明的范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1