核苷酸序列变体的频率分布的确定的制作方法
本公开内容总体涉及分析核苷酸序列样本的领域。更具体地说,涉及在这样一个样本中核苷酸序列变体的频率分布的确定。
背景技术:
在分析核苷酸序列样本(例如脱氧核糖核酸(dna)或核糖核酸(rna))样本时,可能期望能够实现对样本中的核苷酸序列变体的频率分布的准确确定。特别地,可能期望能够实现在样本中围绕感兴趣的特定位点的核苷酸序列变体的频率分布的准确确定。本公开内容的其余部分涉及围绕这样一个感兴趣的特定位点(也称为靶位点或靶位)的核苷酸序列变体。
本文提及的核苷酸序列可具有任何合适的长度并且可包括一个或多个核苷酸序列实体。例如,核苷酸序列可由一个完整基因组、一个基因组的一个或多个染色体、或者一个或多个染色体的一个或多个部分(例如基因)组成。
例如,如果原始核苷酸序列出于某些目的被改变,例如如果特定位点在基因编辑过程中被有意修改(例如与cre组合相比较),则核苷酸序列样本可包括改变的核苷酸序列的集合。在这样一个例子中,能够准确确定有多少被改变的核苷酸序列经历了哪种改变,可能是令人关注的。准确确定在样本中核苷酸序列变体的频率分布将提供这些结果。
用于估计序列变体频率分布的现有方法可能被证明是不准确的;至少在一定程度上是不准确的。
因此,需要用于确定序列变体频率分布的替代方法。优选地,这些方法应当提供更高的准确性,即,改善的准确性。
技术实现要素:
应当强调,当在本说明书中使用时,术语“包括/包含”被用于指定所描述的特征、整数、步骤或组件的存在,但不排除存在或添加一个或多个其他的特征、整数、步骤、组件或其组。如本文所用,单数形式“一”、“一个”和“该”旨在包含复数形式,除非上下文另有明确指示。
应当注意,当本文提及时,术语“确定”在适用的情况下,根据一些实施例,可包括估计。
一些实施例的目的在于解决或减轻、缓解或消除至少一些上述缺点或其他缺点。
根据第一方面,这通过一种用于确定核苷酸序列样本的多个核苷酸序列变体的频率分布的方法实现,其中多个核苷酸序列变体包括核苷酸参考序列,其中每个核苷酸序列变体(其与核苷酸参考序列不同)与核苷酸参考序列在至少一个中间部分上不同,并且与核苷酸参考序列在核苷酸参考序列的至少两个部分上一致。
该方法包括生成序列参考变体的参考变体列表,参考变体列表包括多个核苷酸序列变体的单一核苷酸序列变体,以及,通过对参考变体列表的每个序列参考变体执行多个参考序列读取来生成序列读取数据的参考集,其中每个参考序列读取具有读取起始位置(rss)。
该方法还包括(对于每个rss和每个序列参考变体的中间部分),基于对应参考序列读取至参考变体列表的比对,来确定中间部分是否通过对应参考序列读取可检测。
此外,该方法包括通过对核苷酸序列样本的多个核苷酸序列变体片段中的每一个执行一个或多个样本序列读取来生成序列读取数据的样本集,其中每个样本序列读取对应于参考序列读取并且具有对应的rss。
该方法还包括(对每个rss和每个序列参考变体的中间部分),基于对应样本序列读取至参考变体列表的比对,确定序列读取数据在样本集中的总出现次数。
该方法还包括基于总出现次数来确定多个核苷酸序列变体的频率分布,以及确定对每个rss中间部分是否可检测。
应当注意,本文所述的核苷酸序列变体是围绕核苷酸参考序列的一个感兴趣的的特定位点的核苷酸序列变体。在各种实施例中,本文中的方法可概括为确定和一个以上感兴趣的特定位点相关的频率分布。
通常,rss被定义为和感兴趣的特定位点相关。
在一些实施例中,该方法可包括(对每对序列参考变体,其不包括核苷酸参考序列),对该对中的每个序列参考变体,对其中能够检测中间部分的rss,基于该对中的每个序列参考变体的出现次数来确定在该对序列参考变体之间的相对出现比率,并且基于相对出现比率确定多个核苷酸序列变体的频率分布。
在一些实施例中,该方法还包括定义核苷酸参考序列。
在一些实施例中,中间部分是插入、缺失、修改和转位中的一种或多种。
在一些实施例中,(对rss和序列参考变体的中间部分)确定中间部分是否通过对应参考序列读取可检测,包括通过比对来确定在对应参考序列读取和序列参考变体之间的rss的检测相关性值,当检测相关性值高于检测阈值时,确定中间部分在rss处通过对应参考序列读取可检测,当检测相关性值不高于检测阈值时,确定中间部分在rss处通过对应参考序列读取不可检测。
在一些实施例中,(对rss和序列参考变体的中间部分)确定在样本集中的总出现次数包括通过比对来确定在对应样本序列读取和序列参考变体之间的rss的样本相关性值,当样本相关性值高于出现阈值时,对出现进行计数,当样本相关性值不高于出现阈值时,避免对出现进行计数。
在一些实施例中,该方法还包括接收输入信号,该输入信号指示多个核苷酸序列变体和核苷酸参考序列中的一个或多个。
在一些实施例中,该方法还包括传送报告信号到用户接口,该报告信号指示频率分布。
第二方面是一种包括非暂时性计算机可读介质的计算机程序产品,其上具有包括程序指令的计算机程序。计算机程序可被加载到数据处理单元中,并且被配置成能够当由数据处理单元运行计算机程序时,使得执行根据第一方面的方法。
第三方面是一种用于确定核苷酸序列样本的多个核苷酸序列变体的频率分布的装置,其中多个核苷酸序列变体包括核苷酸参考序列,其中每个核苷酸序列变体(与核苷酸参考序列不同)与核苷酸参考序列在至少一个中间部分上不同,并且与核苷酸参考序列在核苷酸参考序列至少两个部分上一致。
该装置包括控制电路,其被配置成能够使得生成序列参考变体的参考变体列表,参考变体列表包括多个核苷酸序列变体的单一核苷酸序列变体,以及,通过对参考变体列表的每个序列参考变体执行多个参考序列读取来生成序列读取数据的参考集,其中每个参考序列读取具有读取起始位置(rss)。
控制电路还被配置成能够基于对应参考序列读取至参考变体列表的比对(对每个rss和每个序列参考变体的中间部分)来确定中间部分是否通过对应参考序列读取可检测。
此外,控制电路被配置成能够通过对核苷酸序列样本的多个核苷酸序列变体片段中的每一个执行一个或多个样本序列读取来生成序列读取数据的样本集,其中每个样本序列读取对应于参考序列读取,并且具有对应的rss。
控制电路还被配置成能够基于对应样本序列读取至参考变体列表的比对来(对每个rss和每个序列参考变体的中间部分)确定序列读取数据在样本集中的总出现次数。
此外,控制电路被配置成能够使得基于总出现次数确定多个核苷酸序列变体的频率分布,以及对每个rss确定中间部分是否可检测。
在一些实施例中,控制电路可被配置成能够使得,对该对的每个序列参考变体(对每对序列参考变体,不包括核苷酸参考序列),对其中能够检测中间部分的rss,基于确定的该对的每个序列参考变体的出现次数来确定在该对序列参考变体之间的相对出现比率,并且基于相对出现比率来确定多个核苷酸序列变体的频率分布。
第四方面是一种包括第三方面的装置的服务器节点。
在一些实施例中,上述方面中的任意方面可额外地具有与上述对任何其它方面所解释的各种特征中的任意的相同或相应的特征。
一些实施例的优点是,提高了频率分布的精度。这是因为变体的中间部分的相对出现次数仅相对于其中能够检测中间部分的读取起始位置而被确定。
一些实施例的一些其它优点包括:
-更大的变体的量化是可能的(通常为+/-300个碱基对、bp、插入和/或缺失,相比于应用现有技术的方法时的+/-50个碱基对、bp、插入和/或缺失),
-具有只有单侧匹配序列的变体的量化是可能的,例如转位,
-可将所得的变体频率分布提供给统计质量评估(因为所有计算都可能与方差估计一起报告),这转而又提供了再现性的指示(增加了和所报告的测量值相关的用户信心),以及
-能够实现对碎片偏差(及其校正)的描述。
附图说明
以下参考附图对实施例的详细描述中,将显现本公开内容的其他目的、特征和优点。附图不一定按比例绘制,而是着重说明示例性实施例。
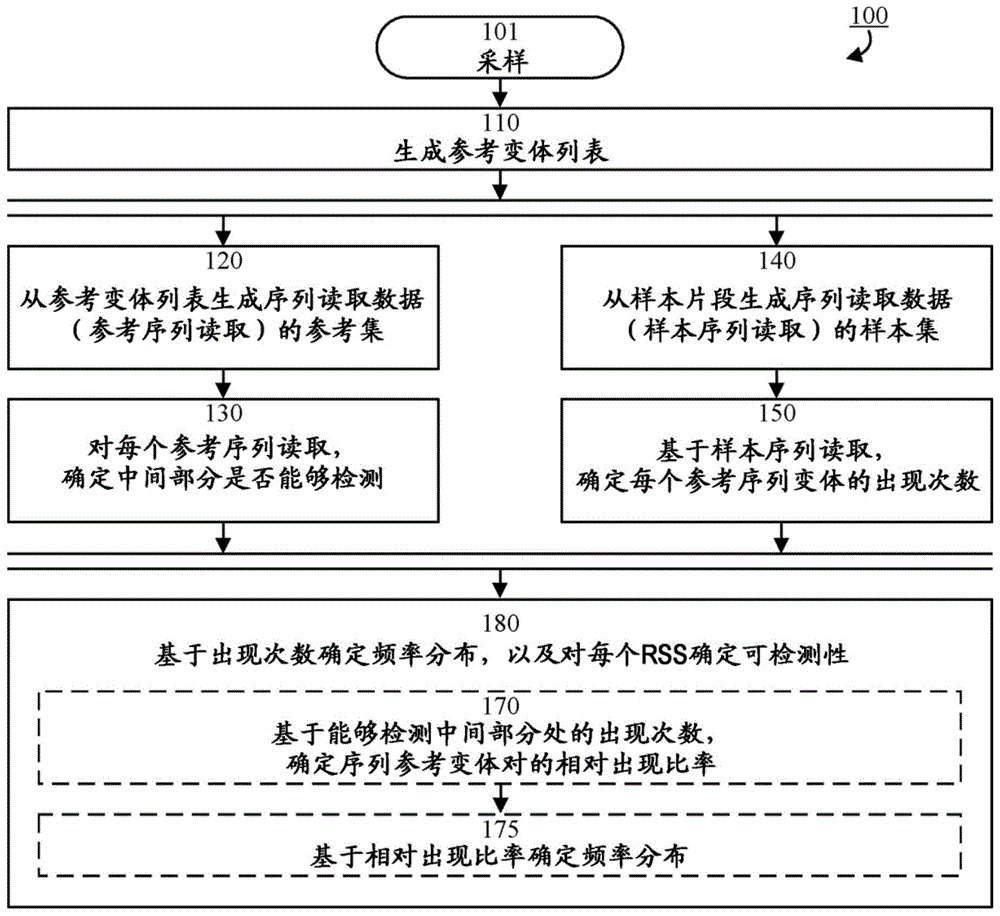
图1是示出根据一些实施例的示例性方法步骤的流程图;
图2是示出根据一些实施例的示例性装置的示意性框图;
图3是示出根据一些实施例的示例性计算机可读介质的示意图;
图4是示出根据一些实施例的示例性方法步骤的流程图;以及
图5和图6是示出根据一些实施例的过程的示例性部分的示意图。
具体实施方式
如上所述,应当强调,当在本说明书中使用时,术语“包括/包含”被用于指定所描述的特征、整数、步骤或组件的存在,但不排除存在或添加一个或多个其他的特征、整数、步骤、组分或其组。如本文所用,单数形式“一”、“一个”和“该”旨在包括复数形式,除非上下文另有明确指示。
下面将参考附图更全面地描述和示例本发明的实施例。然而,本文所公开的解决方案可以多种不同的形式实现,并且不应被解释为限于本文所述的实施例。
下面将描述实施例,其中对于核苷酸序列样本的多个核苷酸序列变体,确定频率分布。
该多个核苷酸序列变体包括核苷酸参考序列。核苷酸参考序列可以被预定义并且作为频率分布的确定过程的输入而被接收。或者,可将核苷酸参考序列确定为频率分布确定过程的初始步骤。例如,在基因编辑中,可将原始核苷酸序列用作核苷酸参考序列,也可将改变的核苷酸序列用作核苷酸参考序列。
多个核苷酸序列变体还包括核苷酸参考序列以外的其他核苷酸序列变体。其他核苷酸序列变体要么与核苷酸参考序列相同,要么与核苷酸参考序列不同。
与核苷酸参考序列不同的每个核苷酸序列变体与核苷酸参考序列在至少一个中间部分上不同,并且与核苷酸参考序列在核苷酸参考序列的至少两个部分上一致。例如,每个中间部分可以是插入、缺失或改变。中间部分的另一个例子是转位。例如,后者可能发生在核苷酸参考序列是全基因组并且其中两个染色体分别被打断并错误地交叉地重新连接时。
图1示出根据一些实施例的用于确定核苷酸序列样本101的多个核苷酸序列变体的频率分布的示例性方法100。在一些实施例中,通过接收输入信号来启动该方法,该输入信号指示多个核苷酸序列变体和/或核苷酸参考序列。
在步骤110中生成序列参考变体的参考变体列表。参考变体列表包括(例如由其组成)多个核苷酸序列变体的单一核苷酸序列变体。例如,参考变体列表可由多个核苷酸序列变体的所有单一核苷酸序列变体或其子集组成。因此,当在多个核苷酸序列变体中存在两个或多个相同的核苷酸序列变体时,通常只将其中一个输入参考变体列表中。
参考变体列表可使用任何合适的方法生成。例如,可通过执行下一代测序(ngs)以产生ngs读取数据,从核苷酸序列样本101生成参考变体列表,并将ngs读取数据相对于核苷酸参考序列重新比对以识别所有中间部分,随后是清理过程,以提供参考变体列表。
在步骤120中,通过对参考变体列表的每个序列参考变体执行多个参考序列读取来生成序列读取数据的参考集。每个参考序列读取都有读取起始位置(rss),其指示在序列参考变体中读取开始的位置。例如,可根据序列参考变体的中间部分的位置来定义读取起始位置。或者,可相对于序列参考变体的另一个位置(例如,开始或结束)来定义读取起始位置。
在典型的实施例中,每个参考序列读取还具有长度和/或方向。例如,长度可定义要在读取中处理的核苷酸序列单元(例如碱基对)的数量,或者与读取中的核苷酸参考序列匹配的核苷酸序列单元(例如碱基对)的数量。对于所有参考序列读取,长度通常可以相等,但不必须相等。方向则可以是正向和反向之一。
在典型示例中,步骤120包括对参考变体列表的每个序列参考变体,执行对序列参考变体的每个可能rss的一个正向参考序列读取,以及,执行对序列参考变体的每个可能rss的一个反向序列读取。
在步骤130中,(对每个rss和每个序列参考变体的中间部分)确定中间部分是否通过对应参考序列读取(即从rss处开始的序列参考变体的参考序列读取)可检测。
通常,只有一些(或没有)参考序列读取延伸覆盖整个中间部分,但是,对延伸覆盖中间部分的起点或终点(变体的一致部分和变体的中间部分之间的桥接点)的参考序列读取,中间部分是可检测的,从而使得可以单一地识别中间部分。
上述确定是基于对应参考序列读取至参考变体列表的比对。可使用任何合适的比对工具进行比对。比对通常可为每个参考序列读取提供检测度量值(例如检测相关性或检测质量分数),其中检测度量值指示参考序列读取与参考变体列表的特定部分(变体和rss)的匹配程度。
此外,可定义检测阈值,检测阈值可以是静态的,也可以是动态可变的。
通常,高检测度量值指示良好的匹配。然后,步骤130可包括在检测度量值高于检测阈值时,确定中间部分在rss处通过对应参考序列读取可检测,否则,确定中间部分在rss处通过对应参考序列读取不可检测。
在其他实施例中,低检测度量值指示良好的匹配,然后,步骤130可包括在检测度量值低于检测阈值时,确定中间部分在rss处通过对应参考序列读取可检测,否则,确定中间部分在rss处通过对应参考序列读取不可检测。
基于比对确定可检测性可称为映射。
在步骤140中,通过对核苷酸序列样本的多个核苷酸序列变体片段中的每一个执行一个或多个样本序列读取来生成序列读取数据的样本集。
核苷酸序列样本的多个核苷酸序列变体片段可以任何合适的方式产生。例如,核苷酸序列样本可在样本制备过程中进行扩增,随后进行ngs文库片段化和标记。
每个样本序列读取对应于参考序列读取,并具有对应rss,对应rss指示在片段中读取开始的位置。如上所述,每个样本序列读取也可具有长度和/或方向。
对于每个核苷酸序列变体片段的一个或多个样本序列读取通常可包括从片段起点的正向读取和/或从片段终点的反向读取。
在步骤150中,对每个rss和每个序列参考变体的中间部分,确定在样本集中的总出现次数,即,对每个rss而言,每个序列参考变体在样本集中的出现次数。
所述确定基于对应样本序列读取至参考变体列表的比对。比对可使用任何合适的比对工具实现。比对通常可为每个样本序列读取提供样本度量值(例如样本相关性或样本质量分数),其中样本度量值指示样本序列读取与参考变体列表的特定部分(变体和rss)的匹配程度。
此外,可定义出现阈值,其可以是静态的,也可以是动态可变的。
通常,高采样度量值指示良好的匹配。然后,步骤150可包括当样本度量值高于出现阈值时,对出现进行计数,否则,避免对出现进行计数。
在其他实施例中,低样本度量值指示良好的匹配,然后,步骤150可包括当样本度量值低于出现阈值时,对出现进行计数,否则,避免对出现进行计数。
基于比对确定总出现次数也可称为映射。
如图1所示,步骤120和130可与步骤140和150并行地执行。然而,这并不意图限制,在其他实施例中,步骤140和150中的一个或多个可以与步骤120和130中的一个或多个顺序执行。例如,根据一些实施例,可以按顺序执行步骤120、130、140和150中的所有步骤。
在步骤180中,基于总出现次数来确定多个核苷酸序列变体的频率分布,并且对每个rss确定中间部分是否可检测。例如,可以通过可选步骤170和175执行该确定,如以下举例说明的。
在步骤170中,对不包括核苷酸参考序列的每对序列参考变体确定相对出现比率。对该对的每个序列参考变体,对其中能够检测中间部分的rss,基于确定的该对的每个序列参考变体的出现次数,确定该比率。
例如,对由第一序列参考变体和第二序列参考变体组成的一对序列参考变体,可以在步骤170中执行以下过程:
-对每个rss检查第一序列参考变体和第二序列参考变体的中间部分是否在rss处可检测。
-如果第一序列参考变体和第二序列参考变体的中间部分中的一个或两个在rss处不可检测,则在该rss处中间相对出现比率是未定义的。
-如果两个都可检测,则对rss计算中间相对出现比率,该中间相对出现比率为第一序列参考变体的总出现次数与第二序列参考变体的总出现次数之间的比率。
-定义该对序列参考变体的相对出现比率为,在其中能够检测两个序列参考变体两者的rss上,中间相对出现比率的(可能加权)平均值。
在步骤175中,基于相对出现比率来确定多个核苷酸序列变体的频率分布。
在一些实施例中,该方法还可包括将报告信号传输到用户接口,该报告信号指示频率分布。
图2示意性地示出根据一些实施例的一种示例性装置210。例如,装置210可被包含在服务器节点中。此外,装置210可适用于使得执行已结合图1描述的一个或多个方法步骤。例如,装置210可适用于执行已结合图1描述的一个或多个方法步骤。
该装置用于确定核苷酸序列样本的多个核苷酸序列变体的频率分布,其中多个核苷酸序列变体包括核苷酸参考序列,其中每个核苷酸变体(与核苷酸参考序列不同)与核苷酸参考序列在至少一个中间部分上不同,与核苷酸参考序列在核苷酸参考序列的至少两个部分上一致。
装置210包括控制电路(cntr;例如一个或多个处理器)200。装置210还可包括存储器电路(mem)220,可连接到控制电路200,或者以其他方式与控制电路200相关联。此外,装置210可包括用户接口电路和/或用户接口装置(ui)230,或与用户接口电路和/或用户接口装置230相关联(例如,相连接)。
控制电路200被配置成能够使得生成序列参考变体的参考变体列表,参考变体列表包括多个核苷酸序列变体的单一核苷酸序列变体(与图1的步骤110类似),通过对参考变体列表的每个序列参考变体执行多个参考序列读取来生成序列读取数据的参考集,其中每个参考序列读取具有读取起始位置(与图1的步骤120类似)。控制电路200还被配置成能够通过对核苷酸序列样本的多个核苷酸序列变体片段中的每一个执行一个或多个样本序列读取来产生序列读取数据的样本集,其中每个样本序列读取对应于一个参考序列读取,并且具有一个对应的读取起始位置(与图1的步骤140类似)。
为此目的,控制电路200可包括生成电路(gen;例如发生器)201,或以其他方式与生成电路201相关联,该生成电路被配置成能够生成参考变体列表、参考集和样本集中的一种或多种。
控制电路200还被配置成能够使得(对每个rss和每个序列参考变体的中间部分),基于对应参考序列读取至参考变体列表的比对,来确定中间部分是否通过对应参考序列读取可检测(与图1的步骤130类似),并基于对应样本序列读取至参考变体列表的比对来确定序列读取数据在样本集中的总出现次数(与图1的步骤150类似)。
为此目的,控制电路200可包括确定电路(det;例如确定器)202,或以其他方式与确定电路202相关联,该确定电路被配置成能够确定中间部分的可检测性和总出现次数中的一种或多种。
控制电路200还被配置成能够使得基于总出现次数确定多个核苷酸序列变体的频率分布,并对每个rss确定中间部分是否可检测(与图1的步骤180类似)。
为此目的,控制电路200可包括频率确定电路(fd;例如确定器)203,或者与频率确定电路203相关联,该频率确定电路被配置成能够确定相对出现比率和频率分布中的一种或多种。
存储器电路220可被配置成能够存储多个核苷酸序列变体、核苷酸参考序列、频率分布以及本文所述的任何中间信息或结果中的一种或多种。
用户接口电路/装置230可被配置成能够从用户处获得包括多个核苷酸序列变体和/或核苷酸参考序列的信息,并通过指示该信息的输入信号将所接收的信息传送至装置210。
在各种实施例中,用户接口电路/装置230还可被配置成能够从用户处获得与执行的分析相关的靶位(靶位点、感兴趣的位点)。
在一些实施例中,用户接口电路/装置230也可被配置成能够从用户处获得多种其他分析偏好参数。这类参数的示例包括:
-一个或多个阈值(例如比对质量的阈值),
-可检测区域边缘之间延伸的安全边界长度,
-归一化的类型和一系列归一化步骤,以及
-其他稳健性的参数。
可替代地或者额外地,用户接口电路/装置230可被配置成能够从装置210接收指示频率分布的报告信号,并处理报告信号以提供用于供用户访问的频率分布的表达。
一般来说,用户接口电路/装置230可由其它合适的接口电路/装置代替、补充或概括。例如,频率分布可不通过用户接口电路230直接提供给用户,而是可提供给其他电路以便利用频率分布。可替代地或者额外地,频率分布可通过用户接口电路230直接提供给用户,并且可经由用户提供给其他电路。
在一些典型示例中,其他电路可以是将频率分布用作参数的计算/处理电路、被配置成能够基于频率分布进行选择的选择电路,以及被配置成能够基于频率分布来调整一种过程的自适应电路中的一种或多种。
通常,频率分布可用于和各种生物应用相关的处理/选择/调整。下文将给出这类应用的几个描述性示例。
在第一个示例中,其目标是对基因编辑研究的应用领域中给定样本的序列变体影响进行分层。在该示例中,输入可以是样本的总变体分布,并且过程可包括确定与原始(第一)参考序列频率相比的非原始参考序列变体的频率,其具有以下指示:
1.在感兴趣的位置处的总体序列变体程度(如果修改百分比高于阈值10%,以20%的低、中、高区间,或其他由用户定义参数)。
2.在该位置处的序列必要性的程度(如果开放读取帧(openreadingframe,简称orf)的中断百分比高于70%,则必要性为负,或者,如果orf的中断百分比低于70%,则必要性为正,以10%的低、中、高程度,或其他用户定义参数)。
3.参考序列变体的多样性程度(如果变体数量是低、中或大的,以[分别地,低于5、介于5至15之间、高于15]的区间,或其他用户定义参数)。
4.是否存在优选参考序列变体(如果相对参考变体频率高于总参考变体频率的10%,或另一用户定义参数)。
5.如果存在优选参考序列变体,则a)为用户打印中间修改的序列给用户,b)一起地,打印完整变体参考序列,并且,如果用户提供了与原始参考序列相关的蛋白质编码序列,则c)提供因中间变体编码序列而新修改的蛋白质编码序列的打印。
在第二个示例中,其目标是对基因编辑研究的应用领域中给定样本的优选参考序列变体进行分层。例如:
1.根据实验设计,沿分析的参考序列来识别关键蛋白质编码区。
2.根据实验设计,沿分析的参考序列来识别关键调节区域。
在该示例中,输入可以是样本或样本组的总变体分布,并且这一过程可包括确定与原始(第一)参考序列频率相比的非原始参考序列变体的频率,其具有以下指示:
1.如果存在一个或多个优选参考序列变体(相对参考变体频率高于总参考变体频率的10%,或另一个用户定义参数),则必须将优选参考序列变体分离出来并打印给用户,对于每个优选参考序列变体指示:
a、优选参考序列变体中间序列和全长度参考序列变体。
b、当前的相对和绝对参考序列变体频率。
c、如果用户提供了参考序列蛋白质编码序列,则打印输出所预测的参考序列变体编码的蛋白质编码序列。
d、如果用户提供了带有监管识别位点注释的原始参考序列,则打印输出所预测的受影响监管位点,其是优选参考序列变体的中间序列所特有的,而并未出现在非优选参考序列变体的中间序列列表中。
2.如果存在一组多个样本,其中一个样本包括优选参考序列变体(相对参考变体频率高于总参考变体频率的10%,或另一个用户定义参数),如果一个或多个样本经受不同的实验设计,则对于每个样本,必须分离优选参考序列变体并打印给用户,对于每个优选参考序列变体指示:
a、优选参考序列变体中间序列和全长度参考序列变体。
b、当前的相对和绝对参考序列变体频率。
c、如果用户提供了参考序列蛋白质编码序列,则打印输出预测的参考序列变体编码的蛋白质编码序列。
d、如果用户提供了带有监管识别位点注释的原始参考序列,则打印输出预测的受影响监管位点,其是优选参考序列变体的中间序列所特有的,而并未出现在非优选参考序列变体的中间序列列表中。
e、如果用户提供了用于样本的比较组从而多个样本包括两组或更多组样本,则用户可以接收不同组中存在的优选参考序列变体类型的打印输出,其具有可能的细分:
i.每组的单一参考序列变体,及其相对和绝对频率。
ii.所有组的共同参考序列变体,及其相对和绝对频率。
iii.各组的特定成对组合的共享参考序列变体,及其相对和绝对频率。
在第三个示例中,目标是对基因编辑研究应用领域中的一组样本的实验设计进行分层。例如:
1.为crispr编辑实验选择最合适的sgrna(例如sgrna1、sgrna2或sgrna3)。
2.选择在基因编辑实验中的最佳编辑时间(例如细胞解冻后立即地编辑、细胞解冻后24小时或细胞解冻后48小时)。
3.选择最佳编辑方式(例如将cas9蛋白与sgrna结合使用,或将cas9mrna与sgrna结合使用)。
在这一示例中,输入可以是能够被串行分析(或在多个计算节点/机器上并行分析)的多用户提交样本的总变体分布,并且该过程可包括在样本之间比较总参考序列变体的分布频率,其具有以下指示:
1.如果样本比较不同的实验条件,则向用户说明样本是否被成功修改(如果修改百分比超过阈值10%,或其他用户定义参数)。
2.实验设计的排序(例如如果一个或多个样本被成功修改,将成功样本从失败样本中分层出来,并按总修改百分比对成功样本进行排序和打印,然后向用户指示最佳实验设计)。成功的实验设计的排序可以根据用户定义参数进行,用户定义参数例如是:总序列变体、序列必要性的程度、变体多样性程度、优选变体数量或优选变体的单一性。
在第四个示例中,其目标是对基因样本临床分析应用领域中的样本的变体程度进行分层。例如:
1.对在用于治疗结果测试或验证的特定参考变体频率的降低进行评估。
2.跟进治疗,以监测预防任何参考序列变体频率的扩展。
在该示例中,输入可以是样本的总变体分布,并且该过程可包括对与原始(第一)参考序列频率相比的非原始参考序列变体频率的评估,其具有以下指示:
1.如果用户没有输入先前所预测的样本变体集,并且如果总样本序列变体程度高于用户定义阈值(例如0.1%或另一个用户定义参数),则向用户通知任何发现的变体在样本中的出现程度(例如,如果低于1%为低程度,1%至5%为中程度,高于5%为高程度,或其他用户定义参数)。
2.如果用户输入先前的或预测的样本变体集,并且与此输入集相比,在分析结束时,任何先前确定的变体的相对变体频率发生了变化(高于或低于用户定义的阈值水平,例如2%),则改变的参考序列变体必须分别打印给用户,对于每个改变的参考序列变体指示:
a、参考序列变体中间序列,并且如果用户提供了参考蛋白质编码序列,则还有变体编码预测的蛋白质编码序列。
b、先前的相对和绝对参考序列变体频率。
c、当前的相对和绝对参考序列变体频率。
3.如果用户输入先前的或预测的样本变体集,并且与此输入集相比,在分析结束时,存在额外或更少的序列变体(高于用户定义阈值水平,例如0.1%),则缺失的参考序列变体和新发现的参考序列变体必须分别打印给用户,对于每个参考序列变体指示:
a、参考序列变体中间序列,并且如果用户提供了参考蛋白质编码序列,则还有变体编码预测的蛋白质编码序列。
b、先前的相对和绝对参考序列变体频率。
c、当前的相对和绝对参考序列变体频率。
在分析期间可能执行其他数据分层点:
1.比对后,对读取进行以下的单独测试,以作为映射的条件:
a、读取长度超出用户指定长度(例如151个碱基)。只有具有相同长度的读取才会被进一步处理。
b、读取比对质量高于用户指定分数(例如10或另一个用户定义参数)。只有具有适当质量分数的读取才会被进一步处理。
c、读取比对匹配由用户指定(例如“151m”或另一个用户定义参数)。只有具有适当比对的读取才会被进一步处理。
2.分析后,对照用户定义阈值参数,评估参考序列变体在样本中的绝对频率,低于该阈值的参考序列变体被视为污染物,无法通过最终质量分析,并将其从用户报告的最终变体集中删除。
根据一些实施例,计算机程序产品包括计算机可读介质,例如通用串行总线(usb)存储器、插入卡、嵌入式驱动器或只读存储器(rom)。图3示出光盘(cd)rom300形式的示例计算机可读介质。计算机可读介质在其上存储了包括程序指令的计算机程序。计算机程序可被加载到数据处理器(proc)320中,例如,该数据处理器(proc)320可被包括在服务器节点310中,或者,可在基于云的实现方式中分布在多个服务器节点中。当计算机程序被加载到数据处理单元时,计算机程序可存储在与数据处理单元相关联的或包含在数据处理单元中的存储器(mem)330中。根据一些实施例,当计算机程序被加载到数据处理单元并由其运行时,可以执行根据例如图1所示或在本文中所描述的其他的一个或多个方法步骤。
图4示出根据一些实施例的一种示例性方法400,并将结合图5和图6的示意图对其进行解释。图4-6可看作是实现图1的方法的一种示例性方式。下面采用dna作为核苷酸序列的示例。
执行和dna样本410、500相关的方法400,以确定dna样本的多个dna变体的频率分布。为了简单起见,仅分别说明三种dna变体(a、b、c)501、502、503。在dna样本中,每个dna变体可能有一种或多种实例。
dna变体(a、b、c)501、502、503相对于参考序列的靶位“0”。靶位是进行分析的位置,并且是参考序列的定义的部分。根据任何合适的过程来生成序列读取数据的样本集(与图1的步骤140类似)。例如,样本制备414(可包括聚合酶链反应pcr)可提供扩增的靶区,并且ngs文库片段可提供ngs片段库460,其经文库标记后称为ngs文库(多个核苷酸序列变体片段)。下一代测序(ngs)464,包括ngs库的正向和反向读取(样本序列读取),可以提供ngs读取数据470、630作为样本集的示例。
多个dna变体包括作为参考dna的dna变体(a)420、501(即核苷酸参考序列)。核苷酸参考序列也可称为第一参考核苷酸序列或第一参考序列。如上所述,如可选步骤412所示的,第一参考核苷酸序列可(例如由用户)预定义或者可被定义为方法400的一部分。
与参考dna不完全相同的每个dna变体(即b、502和c、503)与参考dna在至少一个中间部分504、505上不同,并且与参考dna在参考dna的至少两部分506上一致。如上所述,例如,中间部分举例来说可以是索引、插入、缺失、修改或转位。为了简单起见,中间部分在该示例中用缺失来说明。该方法可包括研究样本的所有中间部分,或仅研究一些定义的中间部分。如可选步骤422所示,包括哪些中间部分可(例如由用户)预定义或可被定义为方法400的一部分。
当确定要包括哪些中间部分时,生成序列参考变体的参考变体列表430、510(与图1的步骤110类似)。参考变体列表通常包括多个dna变体501、502、503中的(一个且仅一个实例)单一dna变体511、512、513;例如,所有单一dna变体或与定义的(例如,在步骤422中定义的)中间部分对应的单一dna变体。如步骤475所示,例如,可基于ngs读取数据,通过重新组装/比对(包括清除)来生成参考变体列表430。
如步骤432所示,通过对参考变体列表的每个序列参考变体执行多个参考序列读取(人工读取数据集生成)来生成序列读取数据的参考集(参考变体读取数据集)440、520。如上所述,每个参考序列读取都有读取起始位置rss521。
如步骤442所示,通过参考序列读取至参考变体列表的比对和映射,对于每个rss和每个序列参考变体的中间部分,基于比对来确定中间部分是否通过对应参考序列读取可检测(与图1的步骤130类似)。该过程产生变体检测限数据集450、540,其指示了对于每个rss和中间部分,中间部分是否在rss处可检测(如图5,540中用“x”标记)或不可检测(如图5,540中示出为空)。图5的540中的粗体框可称为dna变体b的检测限。
如步骤435所示,通过样本序列读取(ngs读取数据)470至参考变体列表430的比对和映射,对于每个rss和每个序列参考变体的中间部分,确定总出现次数(rss计数数据集)480、640(与图1的步骤150类似)。在图6的640中,“1”当然可根据记录的出现次数用任何正整数替换。可选地,如步骤484所示,rss计数数据集可被归一化,从而产生归一化的rss计数数据集490。例如,归一化处理可以覆盖每个rss或每个序列参考变体。
在步骤452中(与图1的步骤170类似),对于每对dna变体(但不包括参考dna)确定相对变体频率分布(相对出现比率)495。对于图5和图6中的示例,b和c之间的相对出现比率可以确定如下:
-rss(-3,+2,+3):因为b和c都不可检测,中间相对出现比率未定义(见540)。
-rss(-2):因为c不可检测,中间相对出现比率未定义(见540)。
-rss(-1,+1):b和c均可检测。中间相对出现比率定义为1/(1+1)=50%。
当然,定义的中间相对出现比率在一般情况下可能有所不同。如果是这样的话,一对dna变体的相对出现比率可被确定为定义的中间相对出现比率在所定义的中间相对出现比率的rss上的平均值(对于每个rss,可能根据出现次数进行加权)。
然后,如步骤497所示,确定频率分布499(与图1的步骤180类似)。如上文所述,首先,对其中能够检测任何非参考序列变体的每个rss,可确定参考dna的中间相对频率。然后,可定义参考dna的相对频率为在rss上的平均值(对于每个rss,可能根据出现次数进行加权)。基于每对dna变体的相对出现比率和参考dna的相对频率,可确定频率分布。
确定频率分布的一种方法的替代性说明还可以描述如下:
-对其中能够检测b和c的每个rss,计算xb/xc,其中xb表示变体序列b的总出现次数,xc表示变体序列c的总出现次数,对不是参考变体序列a的所有变体对重复这一过程。
-对每个特定变体序列i(例如b、c等)的集合(xi/xb,xi/xc…)进行归一化处理。
-在所有归一化集上求取平均值,以给出每个变体的相对频率估计值。
确定频率分布的一种方法的替代性说明还可以描述如下:
1.给定rss计数数据集。
2.对在参考序列变体列表中的每个变体(例如变体a),将变体a的rss矢量除以参考序列变体列表(例如a-z)中的另一个变体(例如变体b)的rss矢量,使得仅当这两个变体(例如a和b)都可检测时才记录除法计算的结果。从该输出矢量中,删除所有不重要的或未定义的结果(通常是:nan、0、1、-inf、+inf)以获得用于a至b的对比的估计点集,并记录集合的平均值。继续对参考序列变体列表中的所有其他变体(例如变体c-z)进行处理,直到列集合(all/a)完整为止。然后对所有变体在第2项(例如b-z)下重复整个过程。
3.对每个列集合(例如all/a、all/b,等等),将各自的列集合除以第一个列集合(all/a),以得到相对的集合比例因子。将列集合除以比例因子得到按比例缩放的列集合。
4.当所有列集合被按比例缩放到第一列集合(all/a)时,继续对经缩放的数据集的每一行取平均值,从而使得每行的平均值是对应变体的相对平均值(第1行是变体a的平均频率,第2行是变体b的平均频率,依此类推)。应当注意,每行的平均值是每行的仅观察到的非零数据点的相对平均值。
5.将最终观察到的变体频率列归一化处理至100%,这样每个变体都被从列的总观测变体计数中给出其相对频率。
6.在归一化过程中,记录每个变体的平均点数量,从而使得
-如果一个变体在其可检测范围内没有任何其他矢量除法伙伴,或者与其他除法伙伴的每个矢量除法对应的估计点非常少(例如,少于5个)
-则将这一变体称为孤立变体。接着,在上述一般的非孤立变体频率的归一化处理之后,仅与原始(第一)参考序列变体的矢量伙伴进行孤立变体的矢量除法计算,使得除法仅在没有其他变体可检测的范围内。
7.然后,如上所述(见第2项),执行孤立变体的除法获得结果,并将结果添加到之前的变体分布中,以补充先前计算的100%的一般非孤立变体频率(例如,非孤立变体100%+新的孤立变体10%=110%),然后重新归一化到一个新的100%的相对参考序列频率分布。
8.在确定相对频率分布的情况下,计算第一原始参考序列矢量,使得对每个rss,第一参考序列计数被转换为第一参考序列频率,如下所示:
9.然后对第一参考序列矢量进行平均,以给出最终的绝对第一参考序列频率,其与之前计算的其余调整后的变体频率分布(见第7项)一起,构成完整的绝对变体频率分布。
如上所述,用于估计序列变体频率分布的现有方法至少在某种程度上是不准确的。例如,许多现有方法都会做出以下一个或多个假设(这些假设可能并不总是正确的):
-基因组dna的pcr扩增是无偏的。
-扩增dna的文库片段(如标记)是一致的。
-由所有读取检测到所有变体类型。
-所有变体类型的变体比对(质量分数)是一致的。
在上述假设下,假设可能是所有事件(插入缺失标记(indel)的检测)具有相同的可能性,因此,将所有检测到的插入和缺失(indels)和所有野生型(wt)读取相加,可以得到在该位点的局部indel分布的平均值。
本文中呈现的实施例根据上述一个或多个假设存在错误的情况做出调整。例如,文库片段通常不是一致的,而是通常会受偏差影响。
本文描述的实施例及其等价物可以在软件或硬件或其组合中实现。实施例可由通用电路执行。通用电路的示例包括数字信号处理器(dsp)、中央处理器(cpu)、协处理器单元、现场可编程门阵列(fpga)和其他可编程硬件。可替代地或额外地,实施例可由专用电路(例如专用集成电路(asic))执行。通用电路和/或专用电路可与该装置相关联或被包含在该装置中,诸如服务器节点中(例如在基于云的实现方式中被分布到多个服务器节点)。
实施例可出现在包括根据本文描述的任何实施例的装置、布置、电路和/或逻辑的电子装置(例如服务器节点)内。可替代地或额外地,电子装置可被配置成能够执行根据本文描述的任何实施例的方法。
一般而言,本文中使用的所有术语应根据其在相关技术领域中的一般含义进行解释,除非在使用该术语的上下文中明确给出不同含义和/或暗示了不同含义。
本文参考了各种实施例。然而,本领域技术人员将认识到在所述实施例基础上的多种变体仍然属于权利要求的范围内。
例如,本文所述的方法实施例通过按一定顺序执行的步骤公开了示例性方法。然而,应当认识到,这些事件的序列可以在不脱离权利要求的范围的情况下以另一顺序发生。此外,一些方法步骤可以并行地执行,即使它们被描述为按顺序执行。因此,本文所公开的任何方法的步骤并不必须按照所公开的确切顺序来执行,除非一个步骤被明确地描述为在另一个步骤的之后或之前,和/或,其中隐含地描述了一个步骤必须在另一个步骤的之后或之前。
与之相同的是,应当理解,在具体的实施例的说明中将功能块划分为功能单元的方式,并不带有任何限制性的意图。相反地,这些划分仅为示例。在此描述为一个单元的功能块可以被分割成两个或多个单元。此外,本文中描述为实现为两个或更多个单元的功能块可以合并成更少的(例如单个)单元。
在适当的情况下,本文所公开的任何实施例的任何特征可应用于任何其他实施例。同样地,任何实施例的任何优点可应用于任何其他实施例,反之亦然。
因此,应当理解,所描述的实施例的细节仅仅是为了说明目的而给出的示例,并且,落入权利要求的范围内的所有变型都将被包括在权利要求中。
- 还没有人留言评论。精彩留言会获得点赞!