制备立他司特的方法与流程
制备立他司特的方法
[0001]
本发明涉及制备式(i)的立他司特(lifitegrast)的方法,立他司特的国际非专利名称是n-[[2-(6-苯并呋喃基羰基)-5,7-二氯-1,2,3,4-四氢-6-异喹啉基]羰基]-3-(甲磺酰基)-l-苯丙氨酸:
[0002]
技术背景
[0003]
用于治疗干燥性角膜结膜炎(kcs)(也称为干眼综合征)的立他司特描述于wo2005044817(sarcode bioscience inc)中。
[0004]
立他司特与整联蛋白lfa-1结合,这是一种在白细胞上发现的细胞表面蛋白,它阻断白细胞与细胞间粘附分子(icam-1)的相互作用,而细胞间粘附分子在眼睛表面的炎症中起重要作用。
[0005]
已经描述了许多合成方法,这些方法涉及中间体的酸性和碱性基团的一系列保护和脱保护,因此,最后的步骤是转化立他司特酯以得到所需产物。所述步骤非常精细,因为如果不加以控制,它会产生低纯度的最终化合物。因此,现有技术寻求开发从酯向酸转化的不同方法,在各种条件下操作以消除所述缺点。
[0006]
us7314938(wo2005044817)中公开的第一种合成方法包括,在步骤n-1中,(s)-苄基-2-氨基-3-(3-(甲磺酰基)苯基)丙酸甲酯和2-(6-苯并呋喃基羰基)-5,7-二氯-1,2,3,4-四氢-6-异喹啉-甲酸之间的缩合反应,和随后所得化合物的碱性水解,得到立他司特。
[0007][0008]
通过将5,7-二氯-1,2,3,4-四氢异喹啉-6-甲酸甲酯的氨基(预先从其叔丁氧基羰基保护基脱保护)与6-苯并呋喃甲酸缩合,得到2-(6-苯并呋喃基羰基)-5,7-二氯-1,2,3,4-四氢-6-异喹啉-甲酸。
[0009]
最后一步是水解甲酯得到羧酸。
[0010][0011]
通过以下方案中报道的合成方法,由(r,r)-(-)-伪麻黄碱甘氨酰胺获得(s)-苄基-2-氨基-3-(3-(甲磺酰基)苯基)丙酸酯盐酸盐:
[0012]
[0013]
在专利申请wo2009139817中,上述合成步骤被颠倒。2-(boc)-5,7-二氯-1,2,3,4-四氢异喹啉-6-甲酸与(s)-苄基-2-氨基-3-(3-(甲磺酰基)苯基)丙酸苄酯缩合,然后除去氨基保护基,以使该化合物与6-苯并呋喃甲酸酰氯缩合。
[0014][0015]
然后进行苄基酯的氢化反应,得到立他司特。
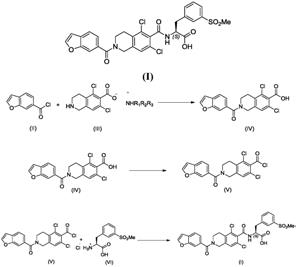
[0016][0017]
wo2011050175特别公开了立他司特酯、尤其是苄酯在酸性/碱性介质中的最后水解步骤。
[0018][0019]
申请wo2014018748旨在通过使用含甲硅烷基的化合物作为立他司特前体酯以便较容易地水解最终化合物来改进最终水解反应。
[0020]
发明描述
[0021]
已令人惊奇地发现,可以避免在立他司特中间体的缩合反应过程中对氨基或酸基的保护,从而分离较少的化合物并避免相应的保护和脱保护步骤。这样,不仅优化了步骤的数目,而且避免了敏感的最终水解反应,结果以良好的产率获得纯净产物。
[0022]
因此,本发明的目的是制备式(i)的立他司特的方法:
[0023][0024]
其包括:
[0025]
a)将式(ii)化合物与式(iii)化合物缩合得到式(iv)化合物
[0026][0027]
其中r1、r2和r3独立地为直链或支链c
1-c6烷基;
[0028]
b)在氯化剂存在下氯化化合物(iv):
[0029][0030]
c)将化合物(v)与氨基酸(vi)缩合得到立他司特(i)
[0031][0032]
d)任选地在极性非质子溶剂和水的混合物中纯化粗品立他司特。
[0033]
步骤a)在合适的溶剂如二甲基甲酰胺、四氢呋喃、甲苯或二氯甲烷,优选二氯甲烷中,在-10℃至40℃的温度下进行。
[0034]
步骤b)在合适的溶剂如四氢呋喃、甲苯或二氯甲烷,优选二氯甲烷中,在0℃至30℃的温度下,在催化量的二甲基甲酰胺存在下进行。以相对于式(iv)化合物为4:1至1.2:1,优选1.5:1的比例使用亚硫酰氯作为氯化剂。
[0035]
步骤c)在合适的溶剂如二甲基甲酰胺、四氢呋喃、甲苯或二氯甲烷,优选二氯甲烷中,在-30℃至40℃,优选-10℃至30℃的温度下进行。
[0036]
涉及纯化粗品立他司特的步骤d)在极性非质子溶剂和水的混合物中进行,优选在乙腈和水的混合物中进行,比例为14:0.5v/v至6:1,有利地在乙腈/水10:1v/v中进行。
[0037]
式(ii)化合物通过使式(vii)的酸与氯化剂反应而获得:
[0038][0039]
反应在常规条件下进行,例如在非质子溶剂中,在碱和/或催化量的二甲基甲酰胺存在下,在20℃至溶剂沸点的温度范围内进行。反应优选在甲苯中在30-60℃的温度下进行。
[0040]
式(iii)化合物通过使化合物(viii)与叔胺nr1r2r3反应而获得,其中基团r1、r2和r3相同或不同,为直链或支链c
1-c4烷基。优选二异丙基乙胺(dipea)。
[0041]
反应优选在极性非质子溶剂如二氯甲烷、氯仿、二甲亚砜和二甲基甲酰胺,优选二氯甲烷中,在溶剂的回流温度下进行。
[0042][0043]
制备立他司特的已知方法包括许多化学步骤。
[0044]
相反,从酰氯(ii)开始的本发明的方法,通过消除大多数已知方法中存在的精细的最终水解步骤,以高收率和较少的步骤数制备了具有高纯度的立他司特。
[0045]
本发明在以下实施例中详细描述。
[0046]
实施例1:化合物(ii)的合成
[0047][0048]
在n2气氛中,将苯并呋喃-6-甲酸(50.0g,0.308mol)、甲苯和催化量的二甲基甲酰胺(dmf)依次加入到反应器中。在搅拌下将悬浮液加热至55
±
5℃。
[0049]
缓慢加入溶于甲苯的socl2(0.370mol),保持温度在55
±
5℃直到转化完全。然后将溶液在真空下浓缩,直到获得几乎为固体的黄色残余物。
[0050]
残余物=55.6g,摩尔收率=定量的。
[0051]1hnmr(300mhz,cdcl3):δ8.32(s,1h),8.01(dd,1h,j1=8.31hz j2=1.34hz),7.87(d,1h,j=2.08hz),7.69(d,1h,j=8.31hz),6.88(br d,1h)。
[0052]
实施例2:化合物(iv)的合成
[0053][0054]
在n2气氛中,将5,7-二氯-1,2,3,4-四氢异喹啉-6-甲酸盐酸盐(30.6g,0.108mol)、ch2cl2和dipea依次加入反应器中。将悬浮液加热至回流2小时至2小时30分钟。
[0055]
将悬浮液冷却至5
±
5℃,缓慢滴加溶解于ch2cl2中的化合物(ii)(0.212mol),保持温度。
[0056]
在室温下搅拌悬浮液直到完全溶解,然后在所述温度下放置16小时。
[0057]
通过加入稀hcl终止反应,并保持温度在20
±
5℃。将所得悬浮液在15-20℃搅拌1-3小时,过滤固体,用ch2cl2和水洗涤。将粗品固体在55-60℃下真空干燥16-20小时。
[0058]
分离的固体=42.0g,摩尔收率=99.0%。
[0059]1hnmr(300mhz,dmso):δ14.06(br.s,1h),8.11(d,1h,j=2.2hz),7.74(m,2h),7.48(br.s,1h),7.35(d,1h,j=8.2hz),7.04(dd,1h,j1=2.2hz j2=0.8hz),4.77(br.s,2h),3.74(br.s,2h),2.85(t,2h,j=5.74hz)。
[0060]
13
cnmr(75mhz,dmso):δ169.91(s),165.98(s),154.13(s),148.14(d),137.86(s),133.70(s),132.64(s),132.11(s),130.38(s),129.11(s),127.45(s),126.45(d),122.49(d),121.86(d),110.81(d),107.28(d),48.92(t),44.33(t),26.89(t)ppm。
[0061]
实施例3:化合物(v)的合成
[0062][0063]
在n2气氛中,将化合物iv(20.0g,0.0513mol)、ch2cl2、催化量的dmf和socl2(0.180mol)在20
±
5℃下依次加入到反应器中。将悬浮液在搅拌下加热至回流,直至转化完全。在真空下将溶液浓缩至残余物,加入甲苯,并将溶液浓缩直至获得固体。
[0064]
固体残余物=21.0g,摩尔产率=定量的。
[0065]
实施例4:化合物(v)的合成
[0066][0067]
在n2气氛中,将化合物iv(20.0g,0.0513mol)、ch2cl2、催化量的dmf和socl2(0.090mol)在20
±
5℃下依次加入到反应器中。将悬浮液在搅拌下加热至回流,直至转化完全。在真空下将溶液浓缩至残余物,加入甲苯,并将溶液浓缩直至获得固体。
[0068]
固体残余物=21.0g,摩尔产率=定量的。
[0069]
实施例5:立他司特(i)的合成
[0070][0071]
在n2气氛中,在20
±
5℃下,将(s)-2-氨基-3-(3-(甲磺酰基)苯基)丙酸盐酸盐(0.0685mol)和ch2cl2依次加入反应器中。在搅拌下将dipea缓慢加入混合物中。冷却混合物,缓慢加入溶解在ch2cl2中的化合物v(20.0g,0.0489mol),保持反应温度在0-5℃。在搅拌下于0-5℃保持反应1小时30分钟至2小时,然后加入meoh。将混合物加热至30
±
5℃,然后冷却至20
±
5℃。加入稀盐酸。分离各相,有机相用水(2
×
120ml)洗涤。将有机相在真空下浓缩至恒重。
[0072]
分离的粗品固体=34.0g,摩尔收率=定量。
[0073]
在n2气氛中,将粗品立他司特(34.0g)在反应器中溶解于ch2cl2。加入硅胶,真空浓缩悬浮液,直到获得残余物。加入80体积的99.0%acoet-acoh混合物,并将悬浮液在20
±
5℃下搅拌30分钟至1小时。过滤二氧化硅并用5体积的99.0%acoet-acoh混合物洗涤。在真空下浓缩溶液直至获得泡沫。
[0074]
将甲乙酮(165ml)加入泡沫中,保持溶液在20
±
5℃下搅拌,加入晶种。将悬浮液在20
±
5℃搅拌12-16小时。过滤固体并用甲乙酮洗涤。
[0075]
将固体在40-45℃干燥至恒重。
[0076]
将粗品固体在甲乙酮(7体积)中回流1小时至1小时30分钟。将悬浮液冷却至20
±
5℃并在4-8小时后过滤。用甲乙酮洗涤固体。
[0077]
将最终分离的固体在40-45℃干燥至恒重。
[0078]
分离的固体=23.2g,摩尔收率=77.0%。
[0079]
所得产物在cukα波长下测量的x射线衍射光谱(图1)与专利us8367701中报道的立他司特a型的x射线衍射光谱(图5,第5页)一致。1hnmr(300mhz,dmso):δ12.87(br.s,1h),9.02(d,1h,j=8.3hz),8.12(d,1h,j=2.2hz),7.88(br.s,1h),7.79-7.55(m,5h),7.49-7.32(br.m,2h),7.05(dd,1h,j1=2.2hz j2=0.9hz),4.80(br.m,3h),3.71(br.s,2h),3.31(dd,1h,j1=14.0hz j2=4.5hz),3.16(s,3h),3.04(dd,1h,j1=14.0hz j2=10.4hz),2.78(br.s,2h)ppm。
[0080]
13
cnmr(75mhz,dmso):δ172.5(s),169.89(s),164.02(s),154.12(s),148.18(d),141.13(s),139.56(s),137.51(s),135.01(s),134.91(d),132.14(s),132.08(s),131.62(s),129.71(d),129.14(s),128.87(s),128.19(d),126.18(d),125.51(d),122.47(d),121.89(d),110.78(d),107.30(d),53.52(d),44.70(t),44.09(q),36.84(t),36.32(t),26.89(t)ppm。
[0081]
实施例6:立他司特(i)的合成
[0082][0083]
在n2气氛中,在20
±
5℃下,向反应器中依次加入(s)-2-氨基-3-(3-(甲磺酰基)苯基)丙酸盐酸盐(0.0587mol)和ch2cl2。加入溶解在ch2cl2中的化合物v(20.0g,0.0489mol),保持反应温度在20
±
5℃。将溶液搅拌5-10分钟并冷却至-5
±
5℃。加入dipea的ch2cl2溶液,并在-5
±
5℃保持5-6小时。加入稀盐酸。在20
±
5℃下分离各相并加入水。将混合物在20
±
5℃下搅拌过夜,并过滤固体。用ch2cl2和h2o洗涤滤饼。将滤饼在50℃下真空干燥至恒重。
[0084]
分离的粗品固体=27.7g,摩尔收率=92.0%。
[0085]
在n2气氛中,在20
±
5℃下,将粗品立他司特(27.7g)和14:1v/v ch3cn/h2o混合物(5体积)依次加入到反应器中。在75
±
5℃搅拌下溶解混合物。在1-2小时内将混合物冷却至20-25℃,并在20-25℃下保持搅拌20-24小时。过滤悬浮液并用h2o(50g)和ch3cn(2
×
30g)洗涤。将湿固体溶解在10:1v/v ch3cn/h2o混合物(估计干物质的4倍体积)中。将混合物与脱色炭一起在75
±
5℃下搅拌溶解,并保持30-40分钟。过滤溶液并用1体积的混合物洗涤。将混合物在1-2小时内冷却至20-25℃,并在搅拌下在20-25℃保持20-24小时。过滤悬浮液并用h2o(2
×
30g)洗涤。
[0086]
分离的粗品固体=21.6g,摩尔收率=76.0%。
[0087]
实施例7:立他司特从ch3cn/h2o中的结晶
[0088][0089]
在n2气氛中,在20
±
5℃下,将立他司特(317g)和10:1v/v ch3cn/h2o混合物(1585ml,5体积)依次加入到反应器中。在75
±
5℃搅拌下溶解混合物。将混合物在1-2小时内冷却至20-25℃,并在搅拌下于20-25℃保持8-12小时。过滤悬浮液,用h2o(2
×
300g)洗涤滤饼两次。在50-55℃下真空干燥固体至恒重。分离的粗固体=280.0g,摩尔收率=88.3%。
[0090]
所得产物在cukα波长下测量的x射线衍射光谱与us8367701中报道的立他司特a型的x射线衍射光谱(图5,第5页)一致。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1