多核苷酸合成方法、试剂盒和系统与流程
[0001]
本发明涉及根据预定的核苷酸序列合成多核苷酸分子的新方法。本发明还涉及合成后组装合成多核苷酸的方法,以及用于进行合成和/或组装方法的系统和试剂盒。
背景技术:
[0002]
存在用于合成和组装多核苷酸分子,特别是dna的两种主要方法。
[0003]
亚磷酰胺化学是通过逐步过程将化学活化的t、c、a或g的单体组装成长度为大约100/150个碱基的寡核苷酸的合成方法。化学反应步骤是高度敏感的,并且条件在完全无水(完全不存在水)、水性氧化和酸性条件之间交替(roy和caruthers,molecules,2013,18,14268-14284)。如果来自先前反应步骤的试剂尚未完全去除,则这将不利于未来的合成步骤。因此,这个合成方法限于产生长度约为100核苷酸的多核苷酸。
[0004]
聚合酶合成方法使用聚合酶使用t、c、a和g三磷酸合成dna模板的互补链。反应条件是水性的和温和的,并且此方法可以用于合成长度为数千个碱基的dna多核苷酸。此方法的主要缺点是单链和双链dna不能通过此方法从头合成,其需要从中制备拷贝的dna模板。(kosuri和church,nature methods,2014,11,499-507)。
[0005]
因此,先前的方法不能用于在没有拷贝的预先存在的模板分子的帮助下从头合成双链dna。
[0006]
本发明人开发了新的方法,通过所述方法可以以逐步的方式从头合成单链和双链多核苷酸分子,而无需复制预先存在的模板分子。这些方法还避免了与亚磷酰胺化学技术相关的极端条件,相反,在中性ph附近的温和水性条件下进行。这样的方法也能使单链或双链多核苷酸分子的从头合成与当前合成方法具有>100聚体的核苷酸长度至完整基因组相比具有潜在的108改进,提供了广泛的在合成生物学可能的应用。
技术实现要素:
[0007]
本发明提供了一种合成具有预定序列的双链多核苷酸的体外方法,该方法包括进行重复的合成循环,其中在每个循环中:
[0008]
(a)通过平端连接反应中连接酶的作用,通过添加预定序列的第一核苷酸来延长双链多核苷酸的第一链;
[0009]
(b)通过核苷酸转移酶或聚合酶添加预定序列的第二核苷酸,使与第一链杂交的双链多核苷酸的第二链延伸;以及
[0010]
(c)然后在裂解位点裂解双链多核苷酸;并且
[0011]
其中每个循环的预定序列的第一和第二核苷酸在裂解后保留在双链多核苷酸中。
[0012]
在任何上述方法中,在每个循环中,第一核苷酸可以是第二核苷酸的配偶体核苷酸,并且其中在合并双链多核苷酸中时,第一和第二核苷酸形成核苷酸对。
[0013]
在任何上述方法中,裂解位点可以由包含通用核苷酸的多核苷酸序列定义。
[0014]
在任何上述方法中,在每个循环中,可以在第二链延伸之前在双链多核苷酸中产
生裂解位点。
[0015]
在任何上述方法中,在步骤(a)期间,通过连接酶的作用,可以将通用核苷酸合并到双链多核苷酸的第一链中以限定裂解位点。
[0016]
在任何上述方法中,第一核苷酸和通用核苷酸可以是多核苷酸连接分子的组分,并且其中,在步骤(a)中,在平端连接反应中,通过裂解酶的作用,多核苷酸连接分子连接至双链多核苷酸,并且其中,在将多核苷酸连接分子连接至双链多核苷酸时,双链多核苷酸的第一链被延伸并且产生了裂解位点。
[0017]
在任何上述方法中,在给定的合成循环中,添加至双链多核苷酸的第二链的该循环的第二核苷酸可包含可逆终止基团,其防止酶进一步延伸,并且其中在将下一循环的第二核苷酸合成的下一循环中添加之前,将可逆终止子基团从该循环并入的第二核苷酸中除去。
[0018]
在任何上述方法中,该方法可以包括执行第一合成循环,包括:
[0019]
(1)提供一种支架多核苷酸,其包含合成链和与其杂交的支持链,其中所述合成链包含引物链部分,并且其中所述支持链是所述双链多核苷酸的第一链,并且合成链是双链多核苷酸的第二链;
[0020]
(2)在平端连接反应中,通过连接酶的作用,将双链多核苷酸连接分子与支架多核苷酸连接,该多核苷酸连接分子包含支持链和与其杂交的辅助链,并且还包含互补连接连接端包括:
[0021]
(i)在支持链中通用核苷酸和预定序列的第一核苷酸;和
[0022]
(ii)辅助链中不可连接的末端核苷酸;
[0023]
其中在连接时,双链多核苷酸的第一链与第一核苷酸一起延伸,并且通过将通用核苷酸合并第一链而产生裂解位点;
[0024]
(3)在所述核苷酸转移酶或聚合酶的作用下,通过合并所述预定序列的第二核苷酸来延伸所述双链支架多核苷酸的所述合成链的所述引物链部分的末端,所述第二核苷酸包括防止通过所述酶进一步延伸的可逆终止子基团,其中,所述第二核苷酸是第一核苷酸的配偶体,并且其中,在合并时,所述第二核苷酸和所述第一核苷酸形成核苷酸对;
[0025]
(4)在裂解位点裂解连接的支架多核苷酸,其中裂解包括裂解支持链并从支架多核苷酸上除去通用核苷酸,以提供包含合并的核苷酸对的裂解的双链支架多核苷酸;和
[0026]
(5)从所述第二核苷酸中移除所述可逆终止子基团;
[0027]
该方法进一步包括进行进一步的合成循环,包括:
[0028]
(6)在平端连接反应中,通过连接酶的作用,将另外的双链多核苷酸连接分子与裂解的支架多核苷酸连接,该多核苷酸连接分子包括支持链和与其杂交的辅助链,并且还包含互补连接端,该连接端包括:
[0029]
(i)在支持链中通用核苷酸和进一步合成循环的第一核苷酸;和
[0030]
(ii)辅助链中不可连接的末端核苷酸;
[0031]
其中在连接时,双链多核苷酸的第一链与进一步的合成循环的第一核苷酸延伸,并且通过将通用核苷酸合并第一链而产生裂解位点;
[0032]
(7)在所述核苷酸转移酶或聚合酶的作用下,通过合并所述进一步的合成循环的所述第二核苷酸来延伸所述双链支架多核苷酸的所述合成链的所述引物链部分的末端,所
述第二核苷酸包括防止通过所述酶进一步延伸的可逆终止子基团,其中,所述进一步的合成循环的所述第二核苷酸是所述进一步的合成循环的所述第一核苷酸的配偶体,并且其中,在合并时,所述进一步的循环的所述第二和第一核苷酸形成另外的核苷酸对;
[0033]
(8)在裂解位点裂解连接的支架多核苷酸,其中裂解包括裂解支持链并从支架多核苷酸中除去通用核苷酸,以提供包含并入的第一和另外的核苷酸对的裂解的双链支架多核苷酸;
[0034]
(9)从所述第二核苷酸中移除所述可逆终止子基团;以及
[0035]
(10)多次重复步骤6至9以提供具有预定核苷酸序列的所述双链多核苷酸。在任何这样的方法中,在任何一个、多个或所有合成循环中,在所述裂解位点处裂解所述连接的支架多核苷酸的步骤之前,可以替代性地将所述可逆终止子基团从所述第二核苷酸中移除。
[0036]
在上述这样的方法中:
[0037]
(a)在第一个循环的连接步骤(步骤2)中以及在所有其他循环的连接步骤中,多核苷酸连接分子的互补连接端可以被构造为使得:
[0038]
i.该循环的预定序列的第一核苷酸是支持链的末端核苷酸,在支持链中位于核苷酸位置n,并与辅助链的末端核苷酸配对;
[0039]
ii.通用核苷酸是支持链的次末端核苷酸,在支持链的核苷酸位置n+1,与辅助链的次末端核苷酸配对;以及
[0040]
iii.辅助链的末端核苷酸是不可连接的核苷酸;
[0041]
其中,位置n是在所述循环的所述预定序列的所述第二核苷酸被合并后与所述第二核苷酸相对的核苷酸位置,并且其中,位置n+1是所述支持链中沿远离所述互补连接端的方向相对于位置n的下一核苷酸位置;并且其中,在连接时,所述多核苷酸连接分子的所述支持链的末端核苷酸连接至与所述合成链的所述引物链部分邻近的所述支架多核苷酸的末端核苷酸,并且在所述辅助链的末端核苷酸与所述合成链的所述引物链部分之间产生单链断裂;
[0042]
(b)在第一循环的延伸步骤(步骤3)和所有其他循环中,将该循环的第二核苷酸合并到与第一链中第一核苷酸相对的第二链中并与之配对;
[0043]
(c)在第一循环的裂解步骤(步骤4)中以及在所有其他循环中,将连接的支架多核苷酸的支持链在位置n+1和n之间裂解,从而从支架多核苷酸释放多核苷酸连接分子并保留该循环的第一核苷酸连接至裂解的支架多核苷酸的第一链,并与该循环的第二核苷酸配对,因此该循环的第一核苷酸在裂解的支架多核苷酸的支持链中所占据的位置定义为在下一合成循环中的核苷酸位置n-1。
[0044]
可替代地,在上述这样的方法中:
[0045]
(a)在第一个循环的连接步骤(步骤2)中以及在所有其他循环的连接步骤中,多核苷酸连接分子的互补连接端可以被构造为使得:
[0046]
i.该循环的预定序列的第一核苷酸是支持链的末端核苷酸,在支持链中位于核苷酸位置n,并与辅助链的末端核苷酸配对;
[0047]
ii.通用核苷酸在支持链中占据核苷酸位置n+2,并与辅助链中的配偶体核苷酸配对;以及
[0048]
iii.辅助链的末端核苷酸是不可连接的核苷酸;
[0049]
其中,位置n是在所述循环的所述预定序列的所述第二核苷酸被合并后与所述第二核苷酸相对的核苷酸位置,并且其中,位置n+2是所述支持链中沿远离所述互补连接端的方向相对于位置n的第二核苷酸位置;并且其中,在连接时,所述多核苷酸连接分子的所述支持链的末端核苷酸连接至与所述合成链的所述引物链部分邻近的所述支架多核苷酸的末端核苷酸,并且在所述辅助链的末端核苷酸与所述合成链的所述引物链部分之间产生单链断裂;
[0050]
(b)在第一循环的延伸步骤(步骤3)和所有其他循环中,将该循环的第二核苷酸合并到与第一链中第一核苷酸相对的第二链中并与之配对;
[0051]
(c)在第一循环的裂解步骤(步骤4)中以及在所有其他循环中,将连接的支架多核苷酸的支持链在位置n+1和n之间裂解,从而从支架多核苷酸释放多核苷酸连接分子并保留该循环的第一核苷酸连接至裂解的支架多核苷酸的第一链,并与该循环的第二核苷酸配对,因此该循环的第一核苷酸在裂解的支架多核苷酸的支持链中所占据的位置定义为在下一合成循环中的核苷酸位置n-1。
[0052]
可替代地,在上述这样的方法中:
[0053]
(a)在第一个循环的连接步骤(步骤2)中以及在所有其他循环的连接步骤中,多核苷酸连接分子的互补连接端可以被构造为使得:
[0054]
i.该循环的预定序列的第一核苷酸是支持链的末端核苷酸,在支持链中位于核苷酸位置n,并与辅助链的末端核苷酸配对;
[0055]
ii.通用核苷酸在支持链中占据核苷酸位置n+2+x,并与辅助链中的配偶体核苷酸配对;以及
[0056]
iii.辅助链的末端核苷酸是不可连接的核苷酸;
[0057]
其中,位置n是在所述循环的所述预定序列的所述第二核苷酸被合并后与所述第二核苷酸相对的核苷酸位置,并且其中,位置n+2是所述支持链中沿远离所述互补连接端的方向相对于位置n的第二核苷酸位置,并且其中,x是沿远离互补连接端的方向相对于位置n+2的核苷酸位置的数目,其中,该数目是1至10或更大的整数;并且其中,在连接时,所述多核苷酸连接分子的所述支持链的末端核苷酸连接至与所述合成链的所述引物链部分邻近的所述支架多核苷酸的末端核苷酸,并且在所述辅助链的末端核苷酸与所述合成链的所述引物链部分之间产生单链断裂;
[0058]
(b)在第一循环的延伸步骤(步骤3)和所有其他循环中,将该循环的第二核苷酸合并到与第一链中第一核苷酸相对的第二链中并与之配对;
[0059]
(c)在第一循环的裂解步骤(步骤4)中以及在所有其他循环中,将连接的支架多核苷酸的支持链在位置n+1和n之间裂解,从而从支架多核苷酸释放多核苷酸连接分子并保留该循环的第一核苷酸连接至裂解的支架多核苷酸的第一链,并与该循环的第二核苷酸配对,因此该循环的第一核苷酸在裂解的支架多核苷酸的支持链中所占据的位置定义为在下一合成循环中的核苷酸位置n-1。
[0060]
在上述任何此类方法中,可以对方法进行修改,以便:
[0061]
(i)在步骤(2)中,多核苷酸连接分子具有互补的连接端,该互补的连接端包含第一循环的预定序列的第一核苷酸,并且还包含第一循环的预定序列的一个或多个其他核苷酸;
[0062]
(ii)在步骤(3)中,通过在所述核苷酸转移酶或聚合酶的作用下合并所述第一循环的所述预定序列的第二核苷酸来延伸所述双链支架多核苷酸的所述合成链的所述引物链部分的末端,并且其中,通过在所述核苷酸转移酶或聚合酶的作用下合并所述第一循环的所述预定序列的一个或多个另外的核苷酸来进一步延伸所述引物链部分的末端,其中,所述第一循环的所述第二核苷酸和所述另外的核苷酸中的每一个包括可逆终止子基团,所述可逆终止子基团阻止通过所述酶进一步延伸,并且其中,在每次进一步延伸之后,在合并下一核苷酸之前,从核苷酸中移除所述可逆终止子基团;
[0063]
(iii)在步骤(4)中,在裂解后,将第一循环的预定序列的第一、第二和其他核苷酸保留在裂解的支架多核苷酸中;
[0064]
(iv)在步骤(6)中,多核苷酸连接分子具有互补的连接端,该互补的连接端包含另外的循环的预定序列的第一核苷酸,并且还包含另外的循环的预定序列的一个或多个另外的核苷酸;
[0065]
(v)在步骤(6)中,通过在所述核苷酸转移酶或聚合酶的作用下合并所述进一步的循环的所述预定序列的第二核苷酸来延伸所述双链支架多核苷酸的所述合成链的所述引物链部分的末端,并且其中,通过在所述核苷酸转移酶或聚合酶的作用下合并所述进一步的循环的所述预定序列的一个或多个另外的核苷酸来进一步延伸所述引物链部分的末端,其中,所述进一步的循环的所述第二核苷酸和所述另外的核苷酸中的每一个包括可逆终止子基团,所述可逆终止子基团阻止通过所述酶进一步延伸,并且其中,在每次进一步延伸之后,在合并下一核苷酸之前,从核苷酸中移除所述可逆终止子基团;
[0066]
(vi)在步骤(8)中,在裂解后,所述另外的循环的预定序列的第一、第二和另外的核苷酸被保留在裂解的支架多核苷酸中。
[0067]
在任何这样的方法中,所述多核苷酸连接分子的所述互补连接端可以被构造为使得在裂解之前的步骤(4)和(8)中,所述通用核苷酸在所述支持链中占据这样的位置:所述位置是所述支持链中沿远离所述互补连接端的方向在所述第一核苷酸和所述另外的核苷酸的核苷酸位置之后的下一核苷酸位置,并且在最后的所述另外的核苷酸所占据的位置与所述通用核苷酸所占据的位置之间裂解所述支持链。替代性地,在任何这样的方法中,所述多核苷酸连接分子的所述互补连接端可以被构造为使得所述多核苷酸连接分子的所述互补连接端被构造为使得在裂解之前的步骤(4)和(8)中,所述通用核苷酸在所述支持链中占据这样的位置:所述位置是所述支持链中沿远离所述互补连接端的方向在所述第一核苷酸和所述另外的核苷酸的核苷酸位置之后的再下一核苷酸位置,并且在所述支持链中的最后的所述另外的核苷酸所占据的位置与所述下一核苷酸所占据的位置之间裂解所述支持链。在任何这样的方法中,可以在裂解位点裂解连接的支架多核苷酸的步骤之前,从最后的核苷酸中去除待结合的另外一个循环的最后一核苷酸的可逆终止子基团。
[0068]
在以上和本文描述的任何方法中,在任何一个、多个或所有合成循环中,与预定序列的第一核苷酸配对的配偶体核苷酸可以是与第一核苷酸互补的核苷酸,优选天然互补。
[0069]
在以上和本文所述的任何方法中,在任何一个,多个或所有合成循环中,可以在步骤(2)和/或(6)之前提供支架多核苷酸,其包含合成链和与其杂交的支持链,并且其中提供的合成链没有辅助链。在任何一个、多个或所有合成循环中,在步骤(2)和/或(6)之前,可以从支架多核苷酸中除去合成链。
[0070]
在以上和本文描述的任何方法中,在任何一个、多个或所有合成循环中,在将双链多核苷酸连接分子连接至裂解的支架多核苷酸的步骤之后并且在将预定核苷酸序列的下一核苷酸合并到支架多核苷酸的合成链中之前,可以从支架多核苷酸中移除合成链的辅助链部分。在任何这样的方法中,可以通过以下步骤从所述支架多核苷酸中移除所述合成链的所述辅助链部分:(i)将所述支架多核苷酸加热至约80℃至约95℃的温度,并从所述支架多核苷酸中分离出所述辅助链部分,(ii)用尿素溶液诸如8m尿素处理所述支架多核苷酸,并且从所述支架多核苷酸中分离出所述辅助链部分,(iii)用甲酰胺或甲酰胺溶液诸如100%甲酰胺处理所述支架多核苷酸,并且从所述支架多核苷酸中分离出所述辅助链部分,或(iv)使所述支架多核苷酸与单链多核苷酸分子接触,所述单链多核苷酸分子包括与所述辅助链部分的序列互补的核苷酸序列区域,从而竞争性地抑制所述辅助链部分与所述支架多核苷酸杂交。
[0071]
在以上和本文所述的任何此类方法中,在相对于通用核苷酸的方向上,在邻近引物的方向上,支架多核苷酸的支持链在通用核苷酸占据的位置和下一核苷酸在支持链中占据的位置之间被裂解合成链的部分,每个裂解步骤可以包括两步裂解过程,其中每个裂解步骤可以包括第一步,该步骤包括去除通用核苷酸从而形成无碱基位点,以及第二步,包括在无碱基位点处裂解支持链。在任何这样的方法中,第一步可以用核苷酸切除酶进行。核苷酸切除酶可为3-甲基腺嘌呤dna糖基化酶。核苷酸切除酶可为人类烷基腺嘌呤dna糖基化酶(haag)或尿嘧啶dna糖基化酶(udg)。在任何这样的方法中,第二步骤可用作为碱的化学品进行。碱可为naoh。在任何这样的方法中,第二步可以用具有无碱基位点裂解活性的有机化学品进行。所述有机化学品可为n,n'-二甲基乙二胺。在任何这样的方法中,第二步可以用具有无碱基位点裂解酶活性的酶进行,如ap核酸内切酶、核酸内切酶iii(nth)或核酸内切酶viii。
[0072]
在上文和本文中所述的任何这样的方法中,其中,沿与所述合成链的所述引物链部分邻近的方向相对于所述通用核苷酸在所述支持链中的由所述通用核苷酸占据的位置与由所述下一核苷酸占据的位置之间裂解所述支架多核苷酸的所述支持链,每个裂解步骤可以包括一个步骤的裂解过程,所述裂解过程包括利用裂解酶移除所述通用核苷酸,其中,所述酶是:核酸内切酶iii、核酸内切酶viii、甲酰胺基嘧啶dna糖基化酶(fpg)或8-氧桥鸟嘌呤dna糖基化酶(hogg1)。
[0073]
在上文和本文中所述的任何这样的方法中,其中,在沿与所述引物链部分邻近的方向相对于所述通用核苷酸的所述支持链中的由所述下一核苷酸占据的位置和沿与所述引物链部分邻近的方向相对于所述通用核苷酸的所述支持链中的由所述第二核苷酸占据的位置之间裂解所述支架多核苷酸的所述支持链,所述裂解步骤可以包括利用酶裂解所述支持链。这种酶可为核酸内切酶v。
[0074]
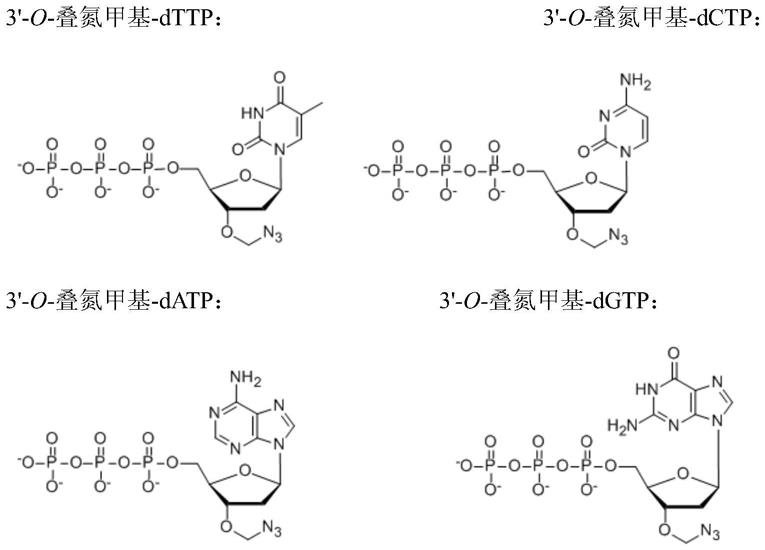
在上文和本文所述的任何方法中,合成的双链多核苷酸的两条链可以是dna链。合成链和支持链可以是dna链。在这种情况下,合并的核苷酸优选是dntp,优选地是包含可逆终止子基团的dntp。在任何此类方法中,包含可逆终止子基团的任何一种或多种或所有合并的核苷酸可包含3
’-
o-烯丙基-dntp或3
’-
o-叠氮甲基-dntp。
[0075]
在上文和本文中所述的任何方法中,所述合成的双链多核苷酸的一条链可以是dna链,并且所述合成的双链多核苷酸的另一条链可以是rna链。合成链可以是rna链,支持
链可以是rna或dna链。在这种情况下,由转移酶或聚合酶合并的核苷酸优选为ntp,优选为包含可逆终止基的ntp。在任何此类方法中,包含可逆终止子基团的任何一种或多种或所有合并的核苷酸可以是3
’-
o-烯丙基-ntp或3
’-
o-叠氮甲基-ntp。
[0076]
在上文和本文中所述的涉及将核苷酸合并到包括dna的合成链中(例如合并一个或多个dntp)的任何方法中,所述酶可以是聚合酶,优选为dna聚合酶,更优选为修饰的dna聚合酶,其具有与未修饰的聚合酶相比增强的合并包括可逆终止子基团的dntp的能力。聚合酶可为来自嗜热球菌(thermococcus)物种9
°
n,优选地物种9
°
n-7的天然dna聚合酶的变体。
[0077]
在上文和本文中所述的涉及将核苷酸合并到包括rna的合成链中(例如合并一个或多个ntp)的任何方法中,所述酶可以是聚合酶,优选为rna聚合酶,诸如t3或t7 rna聚合酶,更优选为修饰的rna聚合酶,其具有与未修饰的聚合酶相比增强的合并包括可逆终止子基团的ntp的能力。
[0078]
在上文和本文所述的任何方法中,合成的双链多核苷酸的第一链可为dna链,并且合成的双链多核苷酸的第二链可为rna链。或者,合成的双链多核苷酸的第一链可为rna链,并且合成的双链多核苷酸的第二链可为dna链。
[0079]
在上文和本文中所述的任何方法中,所述转移酶具有末端转移酶活性,可选地,其中,所述酶是末端核苷酸转移酶、末端脱氧核苷酸转移酶、末端脱氧核苷酸转移酶(tdt)、pol lambda、pol micro或φ29dna聚合酶。
[0080]
在上文和本文所述的任何方法中,从第一/下一核苷酸去除可逆终止子基团的步骤可以用三(羧乙基)膦(tcep)进行。
[0081]
在上文和本文中所述的任何方法中,将双链多核苷酸连接分子连接至所述经裂解的支架多核苷酸的步骤优选地利用连接酶来执行。连接酶可为t3 dna连接酶或t4 dna连接酶。
[0082]
在以上和本文所述的任何方法中,在支架多核苷酸中步骤(1)/(6)中的任何一个、多个或所有合成循环中,合成链包括引物链部分和与其杂交的支持链部分可以通过发夹环连接。
[0083]
在上文和本文中所述的任何方法中,在步骤(2)/(6)中的任何一个、多个或所有合成循环中,在所述多核苷酸连接分子中,所述辅助链和所述支持链的与所述辅助链杂交的部分可以在与所述互补连接端相对的一端通过发夹环连接。
[0084]
在上文和本文中所述的任何方法中,在任何一个、多个或所有合成循环中:
[0085]
a)在支架多核苷酸的步骤(1)/(6)中,包含引物链部分的合成链和与之杂交的支持链的部分通过发夹环连接;以及
[0086]
b)在步骤(2)/(6)中,在所述多核苷酸连接分子中,所述辅助链和所述支持链的与所述辅助链杂交的部分在与所述互补连接端相对的一端通过发夹环连接。
[0087]
在以上和本文所述的任何方法中,可以将多核苷酸连接分子中的至少一个或多个或全部作为单个分子提供,该单个分子包括在与互补连接端相对的一端连接支持链和辅助链的发夹环。在以上和本文描述的任何方法中,每个合成循环的多核苷酸连接分子可以作为单个分子提供,每个分子包括在与互补连接端相对的一端连接支持链和辅助链的发夹环。
[0088]
在上文和本文所述的任何方法中,在步骤(1)和(6)中,可以将包含引物链部分和/或与之杂交的支持链的部分的支架多核苷酸的合成链束缚至共同的表面。包括所述引物链部分和/或所述支持链的与所述引物链部分杂交的部分的所述支架多核苷酸的所述合成链可以包括可裂解的接头,其中,在合成之后,所述接头可以被裂解以从所述表面拆离出所述双链多核苷酸。
[0089]
在以上和本文所述的任何方法中,在步骤(1)和(6)中,合成链的引物链部分和与之杂交的支持链的部分可以通过发夹环连接,并且其中发夹环束缚至表面。
[0090]
在上文和本文中所述的任何方法中,发夹环可以经由可裂解的接头被束缚至表面,其中,在合成之后,所述接头可以被裂解以从所述表面拆离出所述双链多核苷酸。可裂解接头可以是uv可裂解接头。
[0091]
在上文和本文所述的任何方法中,多核苷酸所附着的表面可为微颗粒的表面或平坦表面。
[0092]
在上文和本文所述的任何方法中,多核苷酸所附接的表面可包括凝胶。该表面可以包括聚丙烯酰胺表面,例如约2%的聚丙烯酰胺,优选其中聚丙烯酰胺表面连接到诸如玻璃的固体载体上。
[0093]
在上文和本文所述的任何方法中,包括引物链部分和与其杂交的支持链部分的合成链可以通过一个或多个共价键束缚到共同表面。一个或多个共价键可以在共同表面上的官能团和支架分子上的官能团之间形成,其中支架分子上的官能团可为胺基、硫醇基、硫代磷酸酯基或硫代酰胺基。共同表面上的官能团可以是溴乙酰基,可选地其中溴乙酰基在使用n-(5-溴乙酰基戊基)丙烯酰胺(brapa)衍生的聚丙烯酰胺表面上提供。
[0094]
在上文和本文所述的任何方法中,与上文和本文所述的任何合成循环有关的反应可在微流体系统内以液滴进行。微流体系统可为电润湿系统。微流体系统可为电介质上电润湿系统(ewod)。
[0095]
在上文和本文所述的任何方法中,在合成之后,可以分离双链多核苷酸的链,以提供具有预定义序列的单链多核苷酸。
[0096]
在上文和本文所述的任何方法中,在合成后,扩增双链多核苷酸或其区域,优选通过pcr扩增。
[0097]
本发明还提供了组装具有预定序列的多核苷酸的方法,该方法包括进行上述和本文所述的任何合成方法以合成具有预定序列的第一多核苷酸和具有预定序列的一种或多种另外的多核苷酸并将所述第一多核苷酸和所述一种或多种另外的多核苷酸连接在一起。所述第一多核苷酸和所述一种或多种另外的多核苷酸可优选地包括不同的预定义序列。所述第一多核苷酸和所述一种或多种另外的多核苷酸可为双链的或可为单链的。可首先裂解第一多核苷酸和一种或多种另外的多核苷酸以产生相容的末端,然后例如通过连接将其连接在一起。第一多核苷酸和一种或多种另外的多核苷酸可以在裂解位点被限制酶裂解以产生相容的末端。
[0098]
上文和本文所描述的用于合成具有预定义序列的双链多核苷酸的任何体外方法,和/或上文和本文所描述的用于组装具有预定义序列的多核苷酸的任何体外方法可以在微流体系统内的液滴中进行。在任何这样的方法中,组装方法可以包括组装步骤,其包括提供包括具有预定义序列的第一合成多核苷酸的第一液滴和包括其它一种或多种具有预定义
序列的合成多核苷酸的第二液滴,其中使所述液滴彼此接触并且其中将合成多核苷酸连接在一起,从而组装包括第一核苷酸和其它一种或多种多核苷酸的多核苷酸。在任何这样的方法中,合成步骤可以通过提供多个液滴来进行,每个液滴包括对应于合成循环步骤的反应试剂,并且根据合成循环的步骤将液滴依次递送到支架多核苷酸。在任何这样的方法中,在递送液滴之后并且在递送下一液滴之前,可以进行洗涤步骤以去除过量的反应试剂。在任何这样的方法中,微流体系统可为电润湿系统。在任何这样的方法中,微流体系统可为电介质上电润湿系统(ewod)。在任何这样的方法中,合成和组装步骤可以在同一系统内进行。
[0099]
在如上文和本文中所述的用于合成具有预定序列的双链多核苷酸的任何体外方法中,所述通用核苷酸是肌苷或其类似物、变体或衍生物,并且所述辅助链中的所述通用核苷酸的配偶体核苷酸是胞嘧啶。
[0100]
在相关方面,本发明还提供了延伸双链多核苷酸以合成具有预定序列的双链多核苷酸的体外方法,所述方法包括一个或多个合成循环,其中,在每个合成循环中,在平端连接反应中将通用核苷酸和所述预定序列的第一核苷酸添加至双链支架多核苷酸的第一链,将所述预定序列的第二核苷酸添加至所述支架多核苷酸的相对链,并且在由包括所述通用核苷酸的序列限定的裂解位点处裂解所述支架多核苷酸,其中,在裂解后,所述通用核苷酸从所述支架多核苷酸释放,并且所述第一核苷酸和所述第二核苷酸保留在所述支架多核苷酸中。
[0101]
在相关方面,本发明还提供了通用核苷酸在一种延伸双链多核苷酸以合成具有预定序列的双链多核苷酸的体外方法中的使用,其中,在合成循环中,在平端连接反应中将所述通用核苷酸添加至双链支架多核苷酸以在所述支架多核苷酸中产生多核苷酸裂解位点,并且其中,裂解所述支架多核苷酸以提供所述支架多核苷酸的第一链中的位点用于在下一循环中合并所述预定序列的第一核苷酸以及可选的所述预定序列的一个或多个另外的核苷酸,并且提供所述支架多核苷酸的相对链中的位点用于在所述下一循环中合并所述预定序列的第二核苷酸以及可选的所述预定序列的一个或多个另外的核苷酸。
[0102]
在相关方面,本发明还提供了通用核苷酸在一种合成具有预定序列的双链多核苷酸的体外方法中的使用,其中,在合成循环中使用所述通用核苷酸以在双链支架多核苷酸中产生多核苷酸裂解位点,并且其中,对所述支架多核苷酸的裂解提供了所述支架多核苷酸的每个链中的位点用于合并所述预定序列的一个或多个核苷酸,其中,在每个合成循环中,所述使用包括:提供双链支架多核苷酸,所述双链支架多核苷酸包括合成链和与所述合成链杂交的支持链,其中,所述合成链包括引物链部分;提供双链多核苷酸连接分子,所述双链多核苷酸连接分子包括支持链和与所述支持链杂交的辅助链,并且所述双链多核苷酸连接分子还包括互补连接端,其中,在所述互补连接端的所述辅助链的末端核苷酸包括不可连接的核苷酸,其中,在所述互补连接端的所述支持链的末端核苷酸包括所述预定序列的可连接的第一核苷酸,并且其中,所述支持链包括用于产生多核苷酸裂解位点的通用核苷酸;在平端连接反应中将所述多核苷酸连接分子的支持链连接至所述支架多核苷酸的支持链,因此,利用所述预定序列的所述第一核苷酸延伸所述支架多核苷酸的支持链,并且在所述辅助链与所述合成链的所述引物链部分之间产生单链断裂,并且然后可选地移除所述辅助链;通过聚合酶或转移酶将包括可逆终止子基团的所述预定序列的第二核苷酸添加至所述支架多核苷酸的所述合成链的所述引物链部分的末端;以及在由包括所述通用核苷酸
的序列限定的裂解位点处裂解所述支架多核苷酸的支持链,因此,包括所述通用核苷酸的所述多核苷酸连接分子从所述支架多核苷酸中被移除,并且所述预定序列的所述第一核苷酸和所述第二核苷酸被保持在所述经裂解的支架多核苷酸中;其中,在所述裂解步骤之前或之后,所述可逆终止子基团从所述第二核苷酸中被移除。
[0103]
在任何这样的合成具有预定序列的双链多核苷酸的方法中,所述多核苷酸连接分子的支持链还可以包括与所述预定序列的所述第一核苷酸紧邻的所述预定序列的一个或多个另外的核苷酸,其中,所述预定序列的所述第一核苷酸是所述多核苷酸连接分子的支持链的末端核苷酸并且连接至所述支架多核苷酸的支持链的末端核苷酸;并且其中,在添加所述预定序列的所述第二核苷酸之后,所述方法还包括:通过执行添加包括可逆终止子基团的另外的核苷酸的一个或多个循环,在聚合酶或转移酶的作用下将所述预定序列的一个或多个另外的核苷酸添加至所述合成链的所述引物链部分的末端,并且然后移除所述可逆终止子基团,并且其中,在裂解后,所述预定序列的所述第一核苷酸、所述第二核苷酸和所述另外的核苷酸保留在所述裂解的支架多核苷酸中。
[0104]
通用核苷酸在合成具有预定序列的双链多核苷酸的方法中的这种用途可以使用上文和本文限定和描述的任何特定方法实施。
[0105]
在相关方面,本发明还提供了在相同末端利用预定核苷酸延伸双链多核苷酸分子的每个链的体外方法,所述方法包括:提供双链支架多核苷酸,所述双链支架多核苷酸包括合成链和与所述合成链杂交的支持链,其中,所述合成链包括引物链部分;在平端连接反应中,在连接酶的作用下将所述预定序列的第一核苷酸添加至所述支架多核苷酸的支持链的末端,其中,所述第一核苷酸是双链多核苷酸连接分子的支持链中的末端核苷酸,所述支持链还包括通用核苷酸,其中,所述第一核苷酸连接至所述支架多核苷酸的支持链的末端核苷酸,并且在所述多核苷酸连接分子的所述相对/辅助链与所述合成链的所述引物链部分之间产生单链断裂,以及可选地,移除所述多核苷酸连接分子的所述辅助链;在聚合酶或转移酶的作用下将包括可逆终止子基团的所述预定序列的第二核苷酸添加至所述合成链的所述引物链部分的末端;在由包括所述通用核苷酸的序列限定的裂解位点处裂解所述支架多核苷酸的支持链,因此,包括所述通用核苷酸的所述多核苷酸连接分子从所述支架多核苷酸中被移除,其中,在裂解之后,所述第一核苷酸被保持在所述支持链中,并且所述第二核苷酸被保持在所述引物链部分中;以及从所述第二核苷酸中移除所述可逆终止子基团,其中,所述移除是在裂解之前或之后来执行的。
[0106]
在延伸双链多核苷酸分子的每个链的任何这样的方法中,所述多核苷酸连接分子的支持链还可以包括与所述预定序列的所述第一核苷酸紧邻的所述预定序列的一个或多个另外的核苷酸,其中,所述预定序列的所述第一核苷酸是所述多核苷酸连接分子的支持链的末端核苷酸并且连接至所述支架多核苷酸的支持链的末端核苷酸;并且其中,在添加所述预定序列的所述第二核苷酸之后,所述方法还包括:通过执行添加包括可逆终止子基团的另外的核苷酸的一个或多个循环,在聚合酶或转移酶的作用下将所述预定序列的一个或多个另外的核苷酸添加至所述合成链的所述引物链部分的末端,并且然后移除所述可逆终止子基团,并且其中,在裂解后,所述预定序列的所述第一核苷酸、所述第二核苷酸和所述另外的核苷酸保留在所述裂解的支架多核苷酸中。
[0107]
本发明提供了一种合成具有预定序列的双链多核苷酸的体外方法,所述方法包括
根据前述延伸方法预形成一个或多个延伸循环。
[0108]
在利用预定核苷酸延伸双链多核苷酸分子的每个链的任何这样的方法中,或者在合成具有预定序列的双链多核苷酸的任何这样的方法中,所述方法可以使用上文和本文中定义和描述的任何特定方法来实现。
[0109]
在相关方面,本发明还提供了一种在相同末端利用预定核苷酸延伸双链支架多核苷酸的每个链的循环期间将包括通用核苷酸的多核苷酸连接分子连接至所述双链支架多核苷酸的体外方法,所述方法包括:提供双链支架多核苷酸,所述双链支架多核苷酸包括支持链和与所述支持链杂交的合成链,其中,所述合成链包括引物链部分;以及将双链多核苷酸连接分子连接至所述双链支架多核苷酸,其中,所述多核苷酸连接分子包括支持链和与所述支持链杂交的辅助链,并且所述多核苷酸连接分子还包括互补连接端,其中,在所述互补连接端的所述辅助链的末端核苷酸包括不可连接的核苷酸,其中,在所述互补连接端的所述支持链的末端核苷酸包括所述预定序列的可连接的第一核苷酸,并且其中,所述支持链包括用于产生多核苷酸裂解位点的通用核苷酸,其中,所述连接反应包括:在平端连接反应中将所述多核苷酸连接分子的支持链连接至所述双链支架多核苷酸的支持链,因此,利用所述预定序列的所述第一核苷酸延伸所述支架多核苷酸的支持链,并且在所述辅助链与所述合成链的所述引物链部分之间产生单链断裂,并且然后,可选地移除所述辅助链;所述方法还包括:通过聚合酶或转移酶将包括可逆终止子基团的所述预定序列的第二核苷酸添加至所述支架多核苷酸的所述合成链的所述引物链部分的末端;以及在由包括所述通用核苷酸的序列限定的裂解位点处裂解所述支架多核苷酸的支持链,因此,包括所述通用核苷酸的所述多核苷酸连接分子从所述支架多核苷酸中被移除,并且所述预定序列的所述第一核苷酸和所述第二核苷酸被保持在所述经裂解的支架多核苷酸中;其中,在所述裂解步骤之前或之后从所述第二核苷酸中移除所述可逆终止子基团。
[0110]
在将包括通用核苷酸的多核苷酸连接分子连接至双链支架多核苷酸的任何这样的方法中,所述多核苷酸连接分子的支持链还可以包括与所述预定序列的所述第一核苷酸紧邻的所述预定序列的一个或多个另外的核苷酸,其中,所述预定序列的所述第一核苷酸是所述多核苷酸连接分子的支持链的末端核苷酸并且连接至所述支架多核苷酸的支持链的末端核苷酸;并且其中,在添加所述预定序列的所述第二核苷酸之后,所述方法还包括:通过执行添加包括可逆终止子基团的另外的核苷酸的一个或多个循环,在聚合酶或转移酶的作用下将所述预定序列的一个或多个另外的核苷酸添加至所述合成链的所述引物链部分的末端,并且然后移除所述可逆终止子基团,并且其中,在裂解后,所述预定序列的所述第一核苷酸、所述第二核苷酸和所述另外的核苷酸保留在所述裂解的支架多核苷酸中。
[0111]
本发明提供了一种合成具有预定序列的双链多核苷酸的体外方法,所述方法包括根据前述连接方法预形成一个或多个延伸循环。
[0112]
在合成具有预定序列的双链多核苷酸的循环期间,将包括通用核苷酸的连接多核苷酸连接至双链多核苷酸的任何此类方法中,所述方法可使用上文和本文限定和描述的任何特定方法实施。
[0113]
本发明另外提供了一种用于实施上文和本文中所述的任何合成和/或组装方法的多核苷酸合成系统,所述系统包括:(a)反应区域的阵列,其中,每个反应区域包括至少一个支架多核苷酸;和(b)用于将反应试剂递送至所述反应区域的装置;以及可选地(c)用于从
所述支架多核苷酸中裂解合成的双链多核苷酸的装置。这种系统可进一步包括用于以液滴形式提供反应试剂的装置和用于根据合成循环将液滴输送到支架多核苷酸的装置。
[0114]
本发明进一步提供了与上文和本文所述的任何系统一起使用并且用于实施上文和本文所述的任何合成方法的试剂盒,所述试剂盒包括对应于合成循环的步骤的反应试剂的体积。
[0115]
本发明还提供了制备多核苷酸微阵列的方法,其中所述微阵列包括多个反应区域,每个区域包括一个或多个具有预定义序列的多核苷酸,所述方法包括:
[0116]
a)提供包括多个反应区域的表面,每个区域包括一个或多个双链锚或支架多核苷酸,并且
[0117]
b)在每个反应区域根据上述和本文所述的任何方法进行合成循环,从而在每个区域合成一种或多种具有预定义序列的双链多核苷酸。
[0118]
在此类方法中,在合成后,可分离双链多核苷酸的链以提供微阵列,其中每个区域包括一个或多个具有预定义序列的单链多核苷酸。
附图说明
[0119]
本文提供的和下文描述的相关图显示了使用包含本发明方法的方法的合成循环的一些或所有步骤,以及用于实施方法的方面的方式,例如寡核苷酸、表面、表面附着化学反应、连接子等。这些图以及其所有描述和所有相关方法、试剂和方案仅用于说明呈现而不应解释为限制。
[0120]
相关附图,诸如,例如图6、7、8、9、10、13a、14a、15a等示出了合成循环的一些或所有步骤,包括:合并核苷酸(例如,包括可逆终止子基团的核苷酸)、裂解(例如,将支架多核苷酸裂解成第一部分和第二部分,其中,第一部分包括通用核苷酸,并且第二部分包括合并的核苷酸)、连接(例如,将包括单链部分的多核苷酸构造连接至包括合并的核苷酸的经裂解的支架多核苷酸的第二部分,其中,单链部分包括与合并的核苷酸互补的配偶体核苷酸)和脱保护(例如,从合并的核苷酸中移除可逆终止子基团)。提供这些方法仅用于例示性支持,并不在所要求保护的发明的范围内。图1至5以及图52所示的方法方案是本发明的方法。
[0121]
图1.本发明的示例性方法版本1的方案.
[0122]
示出根据本发明的示例性方法版本1的第一合成循环的方案。
[0123]
该方法包括这样的循环:提供支架多核苷酸;将多核苷酸连接分子连接至支架多核苷酸;合并包括可逆终止子基团或封闭基团的核苷酸;脱保护;以及裂解。
[0124]
该方案示出了提供包括支持链(标记为“a”)和与支持链杂交的合成链(标记为“b”)的支架多核苷酸(101、106)。合成链包含引物链部分(虚线)。与引物链部分邻近的支持链的末端核苷酸包括可连接基团,优选为如图所描绘的末端磷酸基团。引物链部分的末端核苷酸和与其配对的核苷酸被描述为“x”。这两核苷酸可以是任何两核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0125]
该方案显示了多核苷酸连接分子的提供(102、107;图右上方的结构)。多核苷酸连接分子包含辅助链(虚线),与其杂交的支持链和互补的连接端。互补连接端的支持链的末端核苷酸是预定序列的第一核苷酸,并被描述为“a”(腺苷)。互补连接端的辅助链的末端核苷酸描述为“t”(胸腺嘧啶)。互补连接端的辅助链的末端核苷酸包含不可连接的核苷酸。互
补的连接端在支持链中包含通用核苷酸(描述为“un”),并与辅助链中的配偶体核苷酸配对(描述为“x”)。a和t仅是为了举例说明,可以是任何核苷酸或其类似物或衍生物。x可以是任何核苷酸或其类似物或衍生物。配对的核苷酸不必包含天然互补的核苷酸。
[0126]
该方案显示了多核苷酸连接分子(102、107)的支持链与支架多核苷酸的支持链的连接以及在辅助链和引物链部分之间在合成链中的单链断裂(“缺口”)的产生。
[0127]
该方案显示了预定序列的第二核苷酸的合并(103、108)。该核苷酸包含可逆终止基团(三角形),并且仅出于说明目的被描述为“t”(胸腺嘧啶),它可以是任何核苷酸或其类似物或衍生物。
[0128]
该方案显示了脱保护步骤(104、109),其包括从预定序列的第二核苷酸中去除可逆终止子基团。
[0129]
该方案显示了裂解步骤(105、110),其包括在由包含通用核苷酸的序列限定的裂解位点裂解支持链(锯齿状箭头)。裂解释放包含通用核苷酸的多核苷酸连接分子,并引起第一核苷酸和第二核苷酸保留在支架多核苷酸中。在本发明的版本1的合成方法中,在通用核苷酸占据的位置和在接近引物链部分的方向/远离辅助链部分的方向上占据支持链中下一核苷酸位置的核苷酸之间裂解支持链。
[0130]
在本发明的版本1的合成方法中,在每个循环中,预定序列的第一核苷酸和预定序列的第二核苷酸形成核苷酸对。
[0131]
图2.本发明的示例性方法版本2的方案.
[0132]
该方案显示了根据本发明示例性方法版本2的第一合成循环。
[0133]
该方法包括这样的循环:提供支架多核苷酸;将多核苷酸连接分子连接至支架多核苷酸;合并包括可逆终止子基团或封闭基团的核苷酸;脱保护;以及裂解。
[0134]
该方案显示提供了包含支持链(标记为“a”)和与其杂交的合成链(标记为“b”)的支架多核苷酸(201、206)。合成链包含引物链部分(虚线)。与引物链部分邻近的支持链的末端核苷酸包括可连接基团,优选为如图所描绘的末端磷酸基团。引物链部分的末端核苷酸和与其配对的核苷酸被描述为“x”。这两核苷酸可以是任何两核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0135]
该方案显示了多核苷酸连接分子的提供(202、207;图右上方的结构)。多核苷酸连接分子包含辅助链(虚线),与其杂交的支持链和互补的连接端。互补连接端的支持链的末端核苷酸是预定序列的第一核苷酸,并被描述为“a”(腺苷)。互补连接端的辅助链的末端核苷酸描述为“t”(胸腺嘧啶)。互补连接端的辅助链的末端核苷酸包含不可连接的核苷酸。互补的连接端在支持链中包含通用核苷酸(描述为“un”),并与辅助链中的配偶体核苷酸配对(描述为“x”)。在互补连接端的支持链和辅助链的次末端核苷酸都被描述为“x”。a和t仅是为了举例说明,可以是任何核苷酸或其类似物或衍生物。描绘为“x”的核苷酸可以是任何核苷酸或其类似物或衍生物。配对的核苷酸不必包含天然互补的核苷酸。
[0136]
该方案显示了多核苷酸连接分子(202、207)的支持链与支架多核苷酸的支持链的连接以及在辅助链与引物链部分之间在合成链中单链断裂(“缺口”)的产生。
[0137]
该方案显示了预定序列的第二核苷酸的合并(203、208)。该核苷酸包含可逆终止基团(三角形),并且仅出于说明目的被描述为“t”(胸腺嘧啶),它可以是任何核苷酸或其类似物或衍生物。
[0138]
该方案显示了脱保护步骤(204、209),其包括从预定序列的第二核苷酸中去除可逆终止子基团。
[0139]
该方案显示了裂解步骤(205、210),其包括在由包含通用核苷酸的序列限定的裂解位点裂解支持链(锯齿状箭头)。裂解释放包含通用核苷酸的多核苷酸连接分子,并引起第一核苷酸和第二核苷酸保留在支架多核苷酸中。在本发明版本2的合成方法中,在占据相对于通用核苷酸的沿靠近引物链部分/远离辅助链部分的方向的下一核苷酸位置的核苷酸与占据相对于通用核苷酸的沿靠近引物链部分/远离辅助链部分的方向的第二核苷酸位置的核苷酸之间裂解支持链。
[0140]
在本发明版本2的合成方法中,在每个循环中,预定序列的第一核苷酸和预定序列的第二核苷酸形成核苷酸对。
[0141]
图3.显示本发明的示例性方法版本2的变型的方案。
[0142]
该方案显示了根据本发明示例性方法版本2的变体的第一合成循环。
[0143]
该方法包括这样的循环:提供支架多核苷酸;将多核苷酸连接分子连接至支架多核苷酸;合并包括可逆终止子基团或封闭基团的核苷酸;脱保护;以及裂解。
[0144]
该方案显示提供了包含支持链(标记为“a”)和与之杂交的合成链(标记为“b”)的支架多核苷酸(301、306)。合成链包含引物链部分(虚线)。与引物链部分邻近的支持链的末端核苷酸包括可连接基团,优选为如图所描绘的末端磷酸基团。引物链部分的末端核苷酸和与其配对的核苷酸被描述为“x”。这两核苷酸可以是任何两核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0145]
该方案显示了提供多核苷酸连接分子(302、307;图右上方的结构)。多核苷酸连接分子包含辅助链(虚线),与其杂交的支持链和互补的连接端。互补连接端的支持链的末端核苷酸是预定序列的第一核苷酸,并被描述为“a”(腺苷)。互补连接端的辅助链的末端核苷酸描述为“t”(胸腺嘧啶)。互补连接端的辅助链的末端核苷酸包含不可连接的核苷酸。互补的连接端在支持链中包含通用核苷酸(描述为“un”),并与辅助链中的配偶体核苷酸配对(描述为“x”)。在互补的连接端,两核苷酸,表示为“x”,位于支持链的通用核苷酸和末端核苷酸之间,并与辅助链中的配偶体核苷酸配对,也称为“x”。a和t仅是为了举例说明,可以是任何核苷酸或其类似物或衍生物。描绘为“x”的核苷酸可以是任何核苷酸或其类似物或衍生物。配对的核苷酸不必包含天然互补的核苷酸。
[0146]
该方案显示了多核苷酸连接分子(302、307)的支持链与支架多核苷酸的支持链的连接以及辅助链和引物链部分之间在合成链中单链断裂(“缺口”)的产生。
[0147]
该方案显示了预定序列的第二核苷酸的合并(303、308)。该核苷酸包含可逆终止基团(三角形),并且仅出于说明目的被描述为“t”(胸腺嘧啶),它可以是任何核苷酸或其类似物或衍生物。
[0148]
该方案显示了脱保护步骤(304、309),其包括从预定序列的第二核苷酸中去除可逆终止子基团。
[0149]
该方案显示了裂解步骤(305、310),其包括在由包含通用核苷酸的序列限定的裂解位点裂解支持链(锯齿状箭头)。裂解释放包含通用核苷酸的多核苷酸连接分子,并引起第一核苷酸和第二核苷酸保留在支架多核苷酸中。在本发明版本2的合成方法的这些变体中,总是在由预定序列的第一核苷酸占据的位置与由支持链中的沿靠近引物链部分/远离
辅助链部分的方向的下一核苷酸占据的位置之间裂解支持链。
[0150]
在本发明版本2的合成方法的所有这些特定变体中,在每个循环中,预定序列的第一核苷酸和预定序列的第二核苷酸形成核苷酸对。
[0151]
图4.该方案显示了涉及每个循环合并多于两个的核苷酸的本发明示例性方法版本1的变体。
[0152]
该方案显示了根据本发明示例性方法版本1的变体的第一合成循环,该方案包括在连接和合并两个步骤中合并多核苷酸。
[0153]
该方法包括这样的循环:提供支架多核苷酸;将多核苷酸连接分子连接至支架多核苷酸;多个步骤:(a)合并包括可逆终止子基团或封闭基团的核苷酸,随后(b)脱保护;并且然后最终裂解。
[0154]
该方案显示提供了包含支持链(标记为“a”)和与其杂交的合成链(标记为“b”)的支架多核苷酸(401、406)。合成链包含引物链部分(虚线)。与引物链部分邻近的支持链的末端核苷酸包括可连接基团,优选为如图所描绘的末端磷酸基团。引物链部分的末端核苷酸和与其配对的核苷酸被描述为“x”。这两核苷酸可以是任何两核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0155]
该方案显示了提供多核苷酸连接分子(402、407;图右上方的结构)。多核苷酸连接分子包含辅助链(虚线),与其杂交的支持链和互补的连接端。互补连接端的支持链的末端核苷酸是预定序列的第一核苷酸,并被描述为“a”(腺苷)。互补连接端的辅助链的末端核苷酸描述为“t”(胸腺嘧啶)。互补连接端的辅助链的末端核苷酸包含不可连接的核苷酸。支持链的次末端核苷酸是预定序列的另一核苷酸,被描述为“g”(鸟嘌呤),并与辅助链的次末端核苷酸配对,其被描述为“c”(胞嘧啶)。互补的连接端在支持链中包含通用核苷酸(描述为“un”),并与辅助链中的配偶体核苷酸配对(描述为“x”)。a、t、g和c仅是为了举例说明,可以是任何核苷酸或其类似物或衍生物。x可以是任何核苷酸或其类似物或衍生物。配对的核苷酸不必包含天然互补的核苷酸。
[0156]
该方案显示了多核苷酸连接分子(402、407)的支持链与支架多核苷酸的支持链的连接以及辅助链与引物链部分之间在合成链中单链断裂(“缺口”)的产生。
[0157]
该方案显示了预定序列的第二核苷酸的合并(403、408)。该核苷酸包含可逆终止基团(三角形),并且仅出于说明目的被描述为“t”(胸腺嘧啶),它可以是任何核苷酸或其类似物或衍生物。合并后,第二核苷酸与第一核苷酸形成核苷酸对。
[0158]
该方案显示了并入第二核苷酸(404、409)之后的脱保护步骤,该步骤包括从预定序列的第二核苷酸中去除可逆终止子基团。
[0159]
该方案显示了预定序列的另一核苷酸的合并(403'、408')。该核苷酸包含可逆终止基团(三角形),并被描述为“c”(胞嘧啶)。合并后,所述另外的核苷酸与由步骤(2)中的多核苷酸连接分子提供的另外的核苷酸形成核苷酸对,描绘为“g”(鸟嘌呤)。胞嘧啶和鸟嘌呤仅是为了说明而描绘的,这些核苷酸可以是任何核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0160]
该方案显示了并入另外的核苷酸(404'、409')之后的第二脱保护步骤,包括从预定序列的另外的核苷酸中去除可逆终止子基团。
[0161]
该方案显示了裂解步骤(405、410),其包括在由包含通用核苷酸的序列限定的裂
解位点裂解支持链(锯齿状箭头)。裂解释放包含通用核苷酸的多核苷酸连接分子,并引起第一核苷酸、第二核苷酸和其他核苷酸保留在支架多核苷酸中。在本发明版本1的合成方法的该特定变体中,在由通用核苷酸占据的位置与支持链中占据沿靠近引物链部分/远离辅助链部分的方向的下一核苷酸位置的核苷酸之间裂解支持链。
[0162]
在本发明版本1的合成方法的所有这些特定变体中,在每个循环中,预定序列的第一核苷酸和预定序列的第二核苷酸形成核苷酸对,多核苷酸连接分子在步骤(2)中提供的第一其他核苷酸与步骤(3')中合并的第一其他核苷酸形成核苷酸对,依此类推。
[0163]
图5.该方案显示了涉及每个循环合并多于两个的核苷酸的本发明示例性方法版本2的变体。
[0164]
该方案显示了根据本发明示例性方法版本2的变体的第一合成循环,该方案包括在连接和合并两个步骤中合并多核苷酸。
[0165]
该方法包括这样的循环:提供支架多核苷酸;将多核苷酸连接分子连接至支架多核苷酸;多个步骤:(a)合并包括可逆终止子基团或封闭基团的核苷酸,随后(b)脱保护;并且然后最终裂解。
[0166]
该方案显示提供了包含支持链(标记为“a”)和与之杂交的合成链(标记为“b”)的支架多核苷酸(501、506)。合成链包含引物链部分(虚线)。与引物链部分邻近的支持链的末端核苷酸包括可连接基团,优选为如图所描绘的末端磷酸基团。引物链部分的末端核苷酸和与其配对的核苷酸被描述为“x”。这两核苷酸可以是任何两核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0167]
该方案显示了多核苷酸连接分子的提供(502、507;图右上方的结构)。多核苷酸连接分子包含辅助链(虚线)、与其杂交的支持链和互补的连接端。互补连接端的支持链的末端核苷酸是预定序列的第一核苷酸,并被描述为“a”(腺苷)。互补连接端的辅助链的末端核苷酸描述为“t”(胸腺嘧啶)。互补连接端的辅助链的末端核苷酸包含不可连接的核苷酸。支持链的次末端核苷酸是预定序列的另一核苷酸,被描述为“g”(鸟嘌呤),并与辅助链的次末端核苷酸配对,其被描述为“c”(胞嘧啶)。互补的连接端在支持链中包含通用核苷酸(描述为“un”),并与辅助链中的配偶体核苷酸配对(描述为“x”)。描绘为“x”的另外的核苷酸位于支持链中通用核苷酸和预定序列的另外的核苷酸之间。该另外的核苷酸与辅助链中的配偶体核苷酸配对,其也被描述为“x”。a、t、g和c仅是为了举例说明,可以是任何核苷酸或其类似物或衍生物。x可以是任何核苷酸或其类似物或衍生物。配对的核苷酸不必包含天然互补的核苷酸。
[0168]
该方案显示了多核苷酸连接分子(502、507)的支持链与支架多核苷酸的支持链的连接以及辅助链与引物链部分之间在合成链中单链断裂(“缺口”)的产生。
[0169]
该方案显示了预定序列的第二核苷酸的合并(503、508)。该核苷酸包含可逆终止基团(三角形),并且仅出于说明目的被描述为“t”(胸腺嘧啶),它可以是任何核苷酸或其类似物或衍生物。合并后,第二核苷酸与第一核苷酸形成核苷酸对。
[0170]
该方案显示了并入第二核苷酸(504、509)之后的脱保护步骤,该步骤包括从预定序列的第二核苷酸中去除可逆终止子基团。
[0171]
该方案显示了预定序列的另一核苷酸的合并(503'、508')。该核苷酸包含可逆终止基团(三角形),并被描述为“c”(胞嘧啶)。合并后,所述另外的核苷酸与由步骤(2)中的多
核苷酸连接分子提供的另外的核苷酸形成核苷酸对,描绘为“g”(鸟嘌呤)。胞嘧啶和鸟嘌呤仅是为了说明而描绘的,这些核苷酸可以是任何核苷酸或其类似物或衍生物,并且不限于天然互补的核苷酸对。
[0172]
该方案显示了并入另外的核苷酸(504'、509')之后的第二个脱保护步骤,包括从预定序列的另外的核苷酸中去除可逆终止子基团。
[0173]
该方案显示了裂解步骤(505、510),其包括在由包含通用核苷酸的序列限定的裂解位点裂解支持链(锯齿状箭头)。裂解释放包含通用核苷酸的多核苷酸连接分子,并引起第一核苷酸、第二核苷酸和其他核苷酸保留在支架多核苷酸中。在本发明版本2的合成方法中,在占据相对于通用核苷酸的沿靠近引物链部分/远离辅助链部分的方向的下一核苷酸位置的核苷酸与占据相对于通用核苷酸的沿靠近引物链部分/远离辅助链部分的方向的第二核苷酸位置的核苷酸之间裂解支持链。
[0174]
在本发明版本2的合成方法的所有这些特定变体中,在每个循环中,预定序列的第一核苷酸和预定序列的第二核苷酸形成核苷酸对,多核苷酸连接分子在步骤(2)中提供的第一其他核苷酸与步骤(3')中合并的第一其他核苷酸形成核苷酸对,依此类推。
[0175]
图6.示例性方法版本1的方案.
[0176]
该图示出了根据实施例部分的示例性方法版本1的第一合成循环。提供该方法仅用于说明性支持,并且不在所要求保护的发明的范围内。该方法包括提供支架多核苷酸、合并、裂解、连接和脱保护的循环。该方案显示胸腺嘧啶核苷酸在第一个合成循环(101、102)中的合并及其与配偶体腺嘌呤核苷酸(104)相对的配对,以及提供用于下一个合成循环的支架多核苷酸(106)。显示此对仅用于说明目的而不具限制性,取决于所需的预定义序列,此对可以是任何对。核苷酸z可为任何核苷酸。核苷酸x可为任何合适的核苷酸。所述图还示出了对应于第二合成循环的参考标记。
[0177]
图7.示例性方法版本2的方案.
[0178]
该图显示了根据实施例部分的示例性方法版本2的第一合成循环。提供该方法仅用于说明性支持,并且不在所要求保护的发明的范围内。该方法包括提供支架多核苷酸、合并、裂解、连接和脱保护的循环。该方案显示在第一个循环(201、202)中合并胸腺嘧啶核苷酸及其与配偶体腺嘌呤核苷酸相对的配对(204),以及在下一个合成循环中提供包含与胞嘧啶配对的鸟嘌呤的支架多核苷酸(206)。这些对仅用于说明目的而不是限制性的,取决于所需的预定义序列,它们可以是任何对。核苷酸z可为任何核苷酸。核苷酸x可为任何合适的核苷酸。所述图还示出了对应于第二合成循环的参考标记。
[0179]
图8.示例性方法版本3的方案.
[0180]
该图示出了根据实施例部分的示例性方法版本3的第一合成循环。提供该方法仅用于说明性支持,并且不在所要求保护的发明的范围内。该方法包括提供支架多核苷酸、合并、裂解、连接和脱保护的循环。该方案显示在第一个循环(301、302)中合并胸腺嘧啶核苷酸及其与配偶体腺嘌呤核苷酸(304)相对的配对,以及提供用于下一个合成循环的支架多核苷酸(306)。显示此对仅用于说明目的而不具限制性,取决于所需的预定义序列,此对可以是任何对。所述方案还显示胞嘧啶-鸟嘌呤对作为支架多核苷酸的组分并且不是预定义序列的一部分。此对还仅出于说明的目的展示,并且不具有限制性,其可为任何对。核苷酸z可为任何核苷酸。核苷酸x可为任何合适的核苷酸。
[0181]
图9.示例性方法版本4的方案.
[0182]
该图示出了根据实施例部分的示例性方法版本4的第一合成循环。提供该方法仅用于说明性支持,并且不在所要求保护的发明的范围内。该方法包括提供支架多核苷酸、合并、裂解、连接和脱保护的循环。该方案显示在第一个循环(401、402)中合并胸腺嘧啶核苷酸及其与配偶体通用核苷酸相对的配对(404),以及在下一个合成循环中提供包含与胞嘧啶配对的鸟嘌呤的支架多核苷酸(406)。这些对仅用于说明目的而不是限制性的,取决于所需的预定义序列,它们可以是任何对。核苷酸x、y和z可以是任何核苷酸。
[0183]
图10.示例性方法版本5的方案.
[0184]
该图示出了根据实施例部分的示例性方法版本5的第一合成循环。提供该方法仅用于说明性支持,并且不在所要求保护的发明的范围内。该方法包括提供支架多核苷酸、合并、裂解、连接和脱保护的循环。该方案显示在第一个循环(501、502)中合并胸腺嘧啶核苷酸及其与配偶体腺嘌呤核苷酸相对的配对(504),以及在下一个合成循环中提供包含与胞嘧啶配对的鸟嘌呤的支架多核苷酸(506)。所述方案还显示胞嘧啶-鸟嘌呤对(位置n-2)作为支架多核苷酸的组分并且不是预定义序列的一部分。这些对仅用于说明目的而不是限制性的,取决于所需的预定义序列,它们可以是任何对。核苷酸x、y和z可以是任何核苷酸。
[0185]
图11.显示支架多核苷酸表面固定化的方案.
[0186]
方案显示(a至h)支架多核苷酸的可能的示例发夹环构型和其到表面的固定。
[0187]
方案(i和j)显示了用于将多核苷酸连接到表面的表面化学的实例。实例显示了双链实施方案,其中两条链通过发夹连接,但是相同的化学方法可用于附接未连接的双链多核苷酸的一条或两条链。
[0188]
图12.没有辅助链-合并.
[0189]
a)以虚线框突出显示合并步骤的方案。
[0190]
b)对于与肌苷相对合并3
’-
o-修饰的dttp评估dna聚合酶。该图描绘了凝胶,其显示在50℃在mn
2+
离子的存在下通过各种dna聚合酶(bst、deep vent(exo-)、终止子i和终止子ix)进行3
’-
o-改性dttp合并的结果。泳道1:使用bst dna聚合酶合并3
’-
o-烯丙基-dttp。泳道2:使用bst dna聚合酶合并3
’-
o-叠氮甲基-dttp。泳道3:使用deep vent(exo-)dna聚合酶合并3
’-
o-烯丙基-dttp。泳道4:使用deep vent(exo-)dna聚合酶合并3
’-
o-叠氮甲基-dttp。泳道5:使用终止子i dna聚合酶合并3
’-
o-烯丙基-dttp。泳道6:使用终止子i dna聚合酶合并3
’-
o-叠氮甲基-dttp。泳道7:使用终止子ix dna聚合酶合并3
’-
o-烯丙基-dttp。泳道8:使用终止子ix dna聚合酶合并3
’-
o-叠氮甲基-dttp。
[0191]
c)对于与肌苷相对合并3
’-
o-修饰的dttp评估dna聚合酶。使用各种dna聚合酶合并的结果。
[0192]
d)使用终止子ix dna聚合酶评估合并温度。该图描绘了凝胶,其显示了在不同温度下使用终止子ix dna聚合酶在mn
2+
离子存在下与肌苷相对合并3'-修饰的dttp的结果。泳道1:在37℃合并3
’-
o-烯丙基dttp。泳道2:在37℃合并3'-o-叠氮甲基dttp。泳道3:在50℃合并3
’-
o-烯丙基dttp。泳道4:在50℃合并3'-o-叠氮甲基dttp。泳道5:在65℃合并3
’-
o-烯丙基dttp。泳道6:在65℃合并3'-o-叠氮甲基dttp。
[0193]
e)使用终止子ix dna聚合酶评估合并温度。在不同温度下进行的合并的结果。
[0194]
f)使用终止子ix dna聚合酶评估合并时mn
2+
的存在。该图描绘了凝胶,其显示在65
℃相对肌苷合并3'-o-修饰的dttp的结果。泳道s:标准。泳道1:没有mn
2+
离子的3
’-
o-烯丙基-dttp的合并。泳道2:没有mn
2+
离子的3
’-
o-叠氮甲基-dttp的合并。泳道3:在mn
2+
离子存在下合并3
’-
o-烯丙基-dttp。泳道4:在mn
2+
离子存在下合并3
’-
o-叠氮甲基-dttp。
[0195]
g)使用终止子ix dna聚合酶评估合并时mn
2+
的存在。在存在和不存在mn
2+
离子的情况下合并的结果。
[0196]
h)用于研究合并步骤的寡核苷酸。
[0197]
图13.没有辅助链-裂解.
[0198]
a)显示在不存在辅助链的情况下裂解杂交的多核苷酸链的方案。裂解步骤以虚线框突出显示。
[0199]
b)凝胶显示分别在37℃和室温24℃下用haag和0.2m naoh(强碱)裂解寡核苷酸。泳道1.起始寡核苷酸.作为含有两条全长链的正对照的泳道2显示出裂解与未裂解的dna比率为90%:10%的更高产率。包含没有辅助链的裂解反应的泳道3显示裂解与未裂解的dna的比率为10%:90%的低百分比产率。
[0200]
c)凝胶显示寡核苷酸在37℃下用haag和endo viii裂解。作为含有两条全长链的正对照的泳道2显示出裂解与未裂解的dna比率为约90%:10%的更高产率。包含没有辅助链的裂解反应的泳道3显示裂解与未裂解的dna的比率为约7%:93%的低百分比产率。
[0201]
d)用haag/endo viii和haag/化学碱裂解寡核苷酸的总结。
[0202]
e)用于研究裂解步骤的寡核苷酸。
[0203]
图14.没有辅助链-连接.
[0204]
a)显示在不存在辅助链的情况下连接杂交的多核苷酸链的方案。在虚线框中突出显示连接步骤。
[0205]
b)凝胶显示在没有辅助链的情况下在室温(24℃)下用quick t4 dna连接酶连接寡核苷酸。泳道1含有36聚体tamra单链寡核苷酸和18聚体tamra单链寡核苷酸的混合物。这些寡核苷酸用作参考条带。
[0206]
c)用于研究连接步骤的寡核苷酸。
[0207]
图15.用辅助链的版本1化学-合并.
[0208]
a)以虚线框突出显示合并步骤的方案。
[0209]
b)适用于研究合并步骤的寡核苷酸。
[0210]
图16.用辅助链的版本1化学-裂解.
[0211]
a)显示在不存在辅助链的情况下裂解杂交的多核苷酸链的方案。裂解步骤以虚线框突出显示。
[0212]
b)凝胶显示分别在37℃和室温24℃下用haag和0.2m naoh(强碱)裂解寡核苷酸。泳道1.起始寡核苷酸.作为含有两条全长链的正对照的泳道2显示出裂解与未裂解的dna比率为90%:10%的更高产率。包含没有辅助链的裂解反应的泳道3显示裂解与未裂解的dna的比率为10%:90%的低百分比产率。包含与辅助链的裂解反应的泳道4显示裂解与未裂解的dna的比率为50%:50%的相同百分比产率。
[0213]
c)评估核酸内切酶viii裂解无碱基位点。凝胶显示寡核苷酸在37℃下用haag和endo viii裂解。作为含有两条全长链的正对照的泳道2显示出裂解与未裂解的dna比率为约90%:10%的更高产率。包含没有辅助链的裂解反应的泳道3显示裂解与未裂解的dna的
比率为约7%:93%的低百分比产率。包含有辅助链的裂解反应的泳道4显示裂解与未裂解的dna的比率为10%:90%的低百分比产率。
[0214]
d)评估n,n'-二甲基乙二胺裂解无碱基位点。凝胶显示寡核苷酸用haag和100mm n,n'-二甲基乙二胺在37℃下裂解。泳道1.起始寡核苷酸.作为含有两条全长链的正对照的泳道2显示100%裂解的dna。包含有辅助链的裂解反应的泳道3显示裂解与未裂解的dna的比率为90%:10%的较高百分比产率。
[0215]
e)用haag/endo viii、haag/化学碱和haag/替代化学碱裂解寡核苷酸的总结。
[0216]
f)用于研究裂解步骤的寡核苷酸。
[0217]
图17.利用辅助链的版本1化学方法-连接.
[0218]
a)显示在存在辅助链的情况下连接杂交的多核苷酸链的方案。在虚线框中突出显示连接步骤。
[0219]
b)凝胶显示在存在辅助链的情况下在室温(24℃)下用quick t4 dna连接酶连接寡核苷酸。泳道1含有36聚体tamra单链寡核苷酸和18聚体tamra单链寡核苷酸的混合物。这些寡核苷酸用作参考条带。在泳道2中,在20分钟后存在预期条带大小为36聚体的可观察的连接产物。
[0220]
c)凝胶显示在辅助链存在下培育过夜后,在室温(24℃)下用quick t4 dna连接酶连接寡核苷酸。泳道1含有36聚体tamra单链寡核苷酸和18聚体tamra单链寡核苷酸的混合物。这些寡核苷酸作为参考条带。在泳道2中,存在可观察到的完全连接的产物,其预期条带大小为36聚体。
[0221]
d)用于研究连接步骤的寡核苷酸。
[0222]
图18.利用辅助链的版本2化学方法-合并.
[0223]
a)显示合并步骤的方案以橙色虚线框突出显示
[0224]
b)凝胶显示在27℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:起始材料。泳道2:1分钟后合并,转化率为5%。泳道3:2分钟后合并,转化率10%。泳道4:5分钟后合并,转化率20%。泳道5:10分钟后合并,转化率30%。泳道6:20分钟后合并,转化率35%。
[0225]
c)该图描绘了凝胶,其显示在37℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:起始材料。泳道2:1分钟后合并,转化率为30%。泳道3:2分钟后合并,转化率60%。泳道4:5分钟后合并,转化率90%。泳道5:10分钟后合并,转化率90%。泳道6:20分钟后合并,转化率90%。
[0226]
d)凝胶显示在47℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:起始材料。泳道2:1分钟后合并,转化率为30%。泳道3:2分钟后合并,转化率65%。泳道4:5分钟后合并,转化率90%。泳道5:10分钟后合并,转化率90%。泳道6:20分钟后合并,转化率90%。
[0227]
e)凝胶显示在27℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:起始材料。泳道2:1分钟后合并,转化率为70%。泳道3:2分钟后合并,转化率85%。泳道4:5分钟后合并,转化率92%。泳道5:10分钟后合并,转化率96%。泳道6:20分钟后合并,转化率96%。
[0228]
f)凝胶显示在37℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:
起始材料。泳道2:1分钟后合并,转化率为85%。泳道3:2分钟后合并,转化率95%。泳道4:5分钟后合并,转化率96%。泳道5:10分钟后合并,转化率96%。泳道6:20分钟后合并,转化率96%。
[0229]
g)凝胶显示在47℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:起始材料。泳道2:1分钟后合并,转化率为85%。泳道3:2分钟后合并,转化率90%。泳道4:5分钟后合并,转化率96%。泳道5:10分钟后合并,转化率96%。泳道6:20分钟后合并,转化率96%。
[0230]
h)在各种温度和mn
2+
离子存在下合并3
’-
o-叠氮甲基-dttp的总结。
[0231]
i)凝胶显示在37℃在mn
2+
的存在下通过终止子ix dna聚合酶与互补碱基相对3'-o-改性的dntp的合并结果。泳道1:起始材料。泳道2:3
’-
o-叠氮甲基-dttp合并5分钟。泳道3:3
’-
o-叠氮甲基-datp合并5分钟。泳道4:3
’-
o-叠氮甲基-dctp合并5分钟。泳道5:3
’-
o-叠氮甲基-dgtp合并5分钟。
[0232]
j)用于研究合并步骤的寡核苷酸。
[0233]
图19.利用辅助链的版本2化学方法-裂解.
[0234]
a)显示在辅助链存在下裂解杂交的多核苷酸链的方案。裂解步骤以橙色虚线框突出显示。
[0235]
b)凝胶显示用endo v在37℃下裂解寡核苷酸。泳道1.起始寡核苷酸.作为含有两条全长链的正对照的泳道2显示出裂解与未裂解的dna比率为80%:20%的产率。包含没有辅助链的裂解反应的泳道3显示出高得多的裂解的dna产率>99%。包含有辅助链的裂解反应的泳道4也显示出>99%的dna裂解产率。
[0236]
c)核酸内切酶v的裂解研究总结。
[0237]
d)用于研究裂解步骤的寡核苷酸。
[0238]
图20.利用辅助链的版本2化学方法-连接.
[0239]
a)显示在不存在辅助链的情况下连接杂交的多核苷酸链的方案。连接步骤以橙色虚线框突出显示。
[0240]
b)用于研究连接步骤的寡核苷酸。
[0241]
图21.利用辅助链的版本2化学方法-脱保护.
[0242]
a)显示脱保护步骤的方案在橙色虚线框中突出显示。
[0243]
b)该图描绘了凝胶,其显示在合并3
’-
o-叠氮甲基-dttp后通过50mm tcep脱保护3
’-
o-叠氮甲基的结果。泳道1:起始引物
[0244]
泳道2:在mn
2+
存在下合并3
’-
o-叠氮甲基-dttp。泳道3:通过添加所有天然dntp在泳道2中延伸产物。泳道4:通过50mm tcep将泳道2中的产物(0.5μm)脱保护。泳道5:通过添加所有天然dntp在泳道4中延伸产物。
[0245]
c)该图描绘了凝胶,其显示在合并3
’-
o-叠氮甲基-dttp后通过300mm tcep脱保护3
’-
o-叠氮甲基的结果。泳道1:起始引物泳道2:在存在mn
2+
下合并3-o-叠氮甲基-dttp。泳道3:通过添加所有天然dntp在泳道2中延伸产物。泳道4:通过300mm tcep将泳道2中的产物(0.5μm)脱保护。泳道5:通过添加所有天然dntp在泳道4中延伸产物。
[0246]
d)该图描绘了凝胶,其显示在合并3
’-
o-叠氮甲基-dctp后通过50mm tcep脱保护3
’-
o-叠氮甲基的结果。泳道1:起始引物泳道2:在存在mn
2+
下合并3-o-叠氮甲基-dctp。泳道
dna连接酶连接。泳道1含有起始发夹寡核苷酸。1分钟后连接的发夹寡核苷酸的泳道2显示出高产率的连接dna产物,比率为约85%。2分钟后连接的发夹寡核苷酸的泳道3显示出高产率的消化dna,比率为约85%。3分钟后连接的发夹寡核苷酸的泳道4显示出高产率的连接dna产物,比率为约85%。4分钟后连接的发夹寡核苷酸的泳道5显示高产率的连接dna产物,比率为约>85%。
[0267]
c)用于研究连接步骤的发夹寡核苷酸。
[0268]
图25.版本2化学方法-双发夹模型的完整循环.
[0269]
a)显示涉及酶合并、裂解、连接和脱保护步骤的完整循环的方案。
[0270]
b)对于与它的天然对应物相对合并3
’-
o-修饰的dttp评估dna聚合酶。该图描绘了凝胶,其显示在37℃通过终止子ix dna聚合酶合并3'-o-改性dttp的结果。泳道1:起始材料。泳道2:通过终止子ix dna聚合酶合并3
’-
o-叠氮甲基-dttp。泳道3:通过添加所有天然dntp在泳道2中延伸产物。泳道4:通过核酸内切酶v在泳道2中裂解产物。泳道5:通过钝性ta连接酶试剂盒在泳道4中连接产物。
[0271]
c)适用于研究合并步骤的寡核苷酸。
[0272]
图26.版本2化学方法-使用辅助链的单发夹模型的完整循环.
[0273]
a)显示涉及酶合并、裂解、连接和脱保护步骤的完整循环的方案。
[0274]
b)适用于研究合并步骤的寡核苷酸。
[0275]
图27.版本3化学方法-双发夹模型的完整循环.
[0276]
a)显示涉及酶合并、裂解、连接和脱保护步骤的完整循环的方案。
[0277]
b)适用于研究合并步骤的寡核苷酸。
[0278]
图28.版本2化学方法-双发夹模型的完整双循环.
[0279]
a)显示涉及酶促合并、脱保护、裂解和连接步骤的第一个完整循环的方案。
[0280]
b)显示第一个完整循环后的第二个完整循环的方案,涉及酶促合并、脱保护、裂解和连接步骤。
[0281]
c)该图描绘了凝胶,其显示完整双循环实验,包括:合并、脱保护、裂解和连接步骤。
[0282]
泳道1.起始材料。
[0283]
泳道2.用天然dntp延伸起始材料。
[0284]
泳道3.通过终止子ix dna聚合酶合并3
’-
o-叠氮甲基-dttp。
[0285]
泳道4.通过添加所有天然dntp在泳道3中延伸产物。
[0286]
泳道5.tcep在泳道3将产物脱保护。
[0287]
泳道6.通过添加所有天然dntp在泳道5中延伸产物。
[0288]
泳道7.核酸内切酶v在泳道5中裂解产物。
[0289]
泳道8.通过钝性ta连接酶试剂盒在泳道7中连接产物。
[0290]
泳道9.通过λ核酸外切酶裂解泳道8中的产物。
[0291]
泳道10.第二循环的起始材料-与泳道9中的材料相同。
[0292]
泳道11.通过终止子ix dna聚合酶合并3
’-
o-叠氮甲基-dttp。
[0293]
泳道12.通过添加所有天然dntp在泳道11中延伸产物。
[0294]
泳道13.tcep在泳道11将产物脱保护。
[0295]
泳道14.通过添加所有天然dntp在泳道13中延伸产物。
[0296]
泳道15.核酸内切酶v在泳道13中裂解产物。
[0297]
泳道16.通过钝性ta连接酶试剂盒在泳道15中连接产物。
[0298]
d)用于研究的寡核苷酸。
[0299]
图29.
[0300]
显示从根据本文所述方法合成的预定义序列的多核苷酸的支架多核苷酸释放机制的实例。
[0301]
图30.
[0302]
用于根据本发明合成rna的示例性方法的示意图。示例性方法显示在不存在辅助链的情况下合成。
[0303]
图31.
[0304]
用于根据本发明合成rna的示例性方法的示意图。示例性方法显示在辅助链存在下的合成。
[0305]
图32.
[0306]
用于根据本发明合成rna的示例性方法的示意图。示例性方法显示在辅助链存在下的合成。
[0307]
图33.
[0308]
根据具有单发夹模型的合成方法版本2合成dna的示例性方法的第1个完整循环的示意图,包括在合并步骤之前使辅助链变性的步骤。
[0309]
图34.
[0310]
根据具有单发夹模型的合成方法版本2合成dna的示例性方法的第2个完整循环的示意图,包括在合并步骤之前使辅助链变性的步骤。
[0311]
图35.
[0312]
根据具有单发夹模型的合成方法版本2合成dna的示例性方法的第3个完整循环的示意图,包括在合并步骤之前使辅助链变性的步骤。
[0313]
图36.
[0314]
实例9中详述的实验中使用的寡核苷酸。
[0315]
图37.
[0316]
显示对应于实例9中详述的完整三循环实验的反应产物的凝胶。
[0317]
该图描绘了凝胶,其显示了完整的三循环实验的结果,包括:合并、解封闭、裂解和连接步骤。
[0318]
泳道1:起始材料。
[0319]
泳道2.用天然dntp延伸起始材料
[0320]
泳道3:通过终止子x dna聚合酶合并3
’-
o-叠氮甲基-dttp。
[0321]
泳道4:通过添加所有天然dntp延伸泳道3中的产物
[0322]
泳道5:tcep在泳道3中对产物进行解封闭
[0323]
泳道6:通过添加所有天然dntp延伸泳道5中的产物。
[0324]
泳道7:核酸内切酶v对泳道5中的产物的裂解。
[0325]
泳道8:通过t3 dna连接酶在泳道7中连接产物
[0326]
泳道9:第2循环的起始材料-与泳道9中的材料相同。
[0327]
泳道10:通过添加所有天然dntp在泳道9中延伸产物。
[0328]
泳道11:通过终止子x dna聚合酶合并3
’-
o-叠氮甲基-dttp。
[0329]
泳道12:通过添加所有天然dntp在泳道11中延伸产物。
[0330]
泳道13:tcep在泳道11中对产物进行解封闭
[0331]
泳道14:通过添加所有天然dntp在泳道13中延伸产物。
[0332]
泳道15:核酸内切酶v在泳道13中裂解产物
[0333]
泳道16:通过t3 dna连接酶在泳道15中连接产物
[0334]
泳道17:第3循环的起始材料-与泳道16中的材料相同。
[0335]
泳道18:通过添加所有天然dntp在泳道17中延伸产物。
[0336]
泳道19:通过终止子x dna聚合酶合并3
’-
o-叠氮甲基-dttp。
[0337]
泳道20:通过添加所有天然dntp在泳道19中延伸产物。
[0338]
泳道21:tcep在泳道19中对产物进行解封闭
[0339]
泳道22:通过添加所有天然dntp在泳道21中延伸产物。
[0340]
泳道23:核酸内切酶v在泳道21中裂解产物
[0341]
泳道24:通过t3 dna连接酶在泳道23中连接产物
[0342]
图38.
[0343]
来自聚丙烯酰胺凝胶表面的荧光信号合并不同量的brapa,其暴露于fitc-peg-sh和fitc-peg-cooh。
[0344]
图39.
[0345]
测量来自聚丙烯酰胺凝胶表面上的荧光素泳道的荧光信号,其合并不同量的brapa,其暴露于fitc-peg-sh和fitc-peg-cooh。
[0346]
图40.
[0347]
(a)显示没有接头固定在不同的样品上的发夹dna的序列。
[0348]
(b)显示接头固定在不同样品上的发夹dna的序列。
[0349]
图41.
[0350]
来自具有和不具有固定到溴乙酰基官能化的聚丙烯酰胺表面上的接头的发夹dna寡聚物的荧光信号。
[0351]
图42.
[0352]
来自具有和不具有固定到溴乙酰基官能化的聚丙烯酰胺表面上的接头的发夹dna寡聚物的所测量的荧光。
[0353]
图43.
[0354]
在合并三磷酸盐后,来自发夹dna寡聚体的荧光信号,有和没有接头固定在溴乙酰基官能化的聚丙烯酰胺表面上。
[0355]
图44.
[0356]
在合并三磷酸盐之后,测量来自发夹dna寡聚体的荧光,其中有和没有接头固定在溴乙酰基官能化的聚丙烯酰胺表面上。
[0357]
图45.
[0358]
(a)如实例12中详述的每个反应步骤的实验概述和结果。
[0359]
(b)实例12中详述的实验中使用的寡核苷酸。
[0360]
图46.
[0361]
显示在裂解反应之前和之后来自发夹dna寡聚体的荧光信号(实例12)。
[0362]
图47.
[0363]
显示在裂解反应之前和之后测量的来自发夹dna寡聚体的荧光信号(实例12)。
[0364]
图48.
[0365]
显示含肌苷的链和用于连接反应的互补“辅助”链的序列(实例12)。
[0366]
图49.
[0367]
与来自对应于连接反应监测的发夹dna寡聚物的荧光信号有关的结果(实例12)。
[0368]
图50.
[0369]
与来自对应于连接反应监测的发夹dna寡聚物的所测量荧光相关的结果(实例12)。
[0370]
图51.
[0371]
关于通过终止子x dna聚合酶使用本发明方法的合并步骤,例如本发明1和2的合成方法及其变体,通过终止子x dna聚合酶合并3'-o-修饰的-dntp的结果(图1至5和实例13)。
[0372]
图51a提供了引物链(合成链的引物链部分;seq id no:68)和模板链(支持链;seq id no:69)的核酸序列。
[0373]
图51b描绘了示出在37℃下在存在mn2+离子的情况下通过终止子x dna聚合酶合并3
’-
o-修饰的-dntp的结果的凝胶。
[0374]
泳道1:起始寡核苷酸。
[0375]
泳道2:合并3'-o-叠氮甲基-dttp(效率>99%)
[0376]
泳道3:3'-o-叠氮甲基-datp的合并(效率>99%)。
[0377]
泳道4:合并3'-o-叠氮甲基-dctp(效率>90%)。
[0378]
泳道5:合并3'-o-叠氮甲基-dgtp(效率>99%)。
[0379]
添加后,新添加的3'-o-修饰的-dntp在引物链部分中占据位置n。引物链部分中的下一核苷酸位置称为n-1。
[0380]
图52.
[0381]
该图显示了描述实例14中所述的dna合成反应循环的方案。
[0382]
图53.
[0383]
该图显示了在实例14中描述的实验中使用的寡核苷酸。
[0384]
图54.如实例14中所述的连接实验的结果。
[0385]
该图显示了凝胶的照片,该凝胶证明了在用作通用核苷酸的由2-脱氧尿苷限定的裂解位点处裂解发夹支架多核苷酸的步骤的结果,然后将包含2-脱氧尿苷的多核苷酸连接分子连接至裂解的支架多核苷酸。凝胶的泳道如下:
[0386]
泳道1:起始发夹支架多核苷酸。
[0387]
泳道2:使用尿嘧啶dna糖基化酶和核酸内切酶viii的混合物裂解发夹式支架多核苷酸。
[0388]
泳道3:裂解的发夹支架多核苷酸连接至多核苷酸连接分子。
[0389]
附图的解释。
[0390]
图11、12a、13a、14a、15a、16a、17a、18a、19a、20a、21a、22a、23a、24a、25a、26a、27a、28a、28b、29、30、31、32、33、34和35中所描绘的结构将与图6、7、8、9和10中所描绘的结构一致地解释。因此,在这些图中,双链支架多核苷酸分子的每条左手链均与支持链相关(对应于图6至10中的链“a”);双链支架多核苷酸分子的每条右手链均与合成链相关(对应于图6至10中的链“b”);所有支架多核苷酸分子包含较低的合成链,其对应于包含引物链部分的链(对应于图6至图10的链“b”的实线和点线);在合并新核苷酸之前,显示出了某些支架多核苷酸分子(例如图15a和23a),其具有上部合成链,该上部合成链对应于包含辅助链部分的链(对应于图6至10中的链“b”的虚线);某些支架多核苷酸分子(例如在图12a、13a和14a中)显示为没有辅助链部分(对应于在图6至10中不存在链“b”的虚线);在连接步骤之后,示出了某些支架多核苷酸分子(例如,图33、34和35),其上部合成链对应于包含辅助链部分的链(对应于图6至10中的链“b”的虚线),并且其中在下一合成循环中合并新核苷酸之前移除辅助链部分。
[0391]
此外,在这些图中,在适当的位置,每个新核苷酸均显示为与可逆终止子基团结合在一起,标记为rtntp,并描绘为小圆形结构(对应于图6至10中的小三角形结构),并且末端磷酸基团标记为“p”且描绘为小的椭圆形结构。
[0392]
图11c、11d、11g、11h、22a、23a、24a、25a、27a、28a、28b和29展示支架多核苷酸分子,其中包括辅助链部分和支持链的链通过发夹环连接。图11b、22a、23a、24a、25a、26a、27a、28a、28b、29、33、34和35展示支架多核苷酸分子,其中包括引物链部分和支持链的链通过发夹环连接。
[0393]
例如图27a和28a的图显示了支架多核苷酸分子,其中包括辅助链部分(右上链)和支持链(左上链)的链通过发夹环连接,并且在同一分子中,通过发夹环连接引物引物链部分(右下链)和支持链(左下链)的链。
具体实施方式
[0394]
本发明提供了根据预定的核苷酸序列从头合成多核苷酸分子的方法。合成的多核苷酸优选为dna,并且优选为双链多核苷酸分子。与现有的合成方法相比,本发明提供了优点。例如,所有反应步骤可以在温和ph的含水条件下进行,不需要广泛的保护和脱保护程序。此外,合成不依赖于复制包括预定的核苷酸序列的预先存在的模板链。
[0395]
本发明人已经确定,如本文所定义的通用核苷酸的使用允许在合成区域内创建多核苷酸裂解位点,其促进合成的裂解和重复循环。本发明提供了用于合成多核苷酸和用于组装包括此类合成多核苷酸的大片段的通用方法。
[0396]
通过参考包括本发明的两个示例性方法版本及其某些变体(图1至图5)的示例性方法,本文将更一般性地描述本发明的合成方法的某些实施方案。应当理解,包括本发明的示例性方法版本及其变体在内的所有示例性方法均不旨在限制本发明。本发明提供了合成具有预定序列的双链多核苷酸分子的体外方法,该方法包括执行合成循环,其中在每个循环中,通过合并预定序列的第一核苷酸来延伸第一多核苷酸链,并且然后通过合并预定序列的第二核苷酸来延伸与第一链杂交的第二多核苷酸链。优选地,所述方法用于合成dna。提供本文描述的具体方法作为本发明的实施例。
[0397]
反应条件
[0398]
在一个方面,本发明提供了合成具有预定义序列的双链多核苷酸的方法。
[0399]
在一些实施方案中,合成在适合于双链多核苷酸内的核苷酸杂交的条件下进行。通常在允许核苷酸与互补核苷酸杂交的条件下使多核苷酸与试剂接触。允许杂交的条件是本领域公知的(例如,sambrook等人,2001,molecular cloning:a laboratory manual,3rd edition,cold spring harbour laboratory press;和current protocols in molecular biology,greene publishing and wiley-lnterscience,new york(1995))。
[0400]
可以在合适的条件下将核苷酸合并到多核苷酸中,例如使用聚合酶(例如,终止子ix聚合酶)或末端脱氧核苷酸转移酶(tdt)或其功能变体,以在存在适当的缓冲溶液的情况下在适当的温度下(例如,约65℃)合并修饰的核苷酸(例如,3
’-
o-修饰-dntp)。在一个实施方案中,缓冲溶液可以包括2mm tris-hcl、1mm(nh4)2so4、1mm kcl、0.2mm mgso4和0.01%x-100。
[0401]
多核苷酸的裂解可以在合适的条件下进行,例如在合适的缓冲溶液存在下,在与酶相容的温度(例如37℃)下使用多核苷酸裂解酶(例如核酸内切酶)。在一个实施方案中,缓冲溶液可包括5mm乙酸钾、2mm tris-乙酸盐、1mm乙酸镁和0.1mm dtt。
[0402]
多核苷酸的连接可以在合适的条件下进行,例如在合适的缓冲溶液存在下,在与酶相容的温度(例如室温)下使用连接酶(例如t4 dna连接酶)。在一个实施方案中,缓冲溶液可以包括4.4mm tris-hcl、7mm mgcl2、0.7mm二硫苏糖醇、0.7mm atp、5%聚乙二醇(peg6000)。
[0403]
脱保护可以在合适的条件下进行,例如使用还原剂(例如tcep)。例如,可以使用tris缓冲液中的tcep(例如终浓度为300mm)进行脱保护。
[0404]
锚多核苷酸和支架多核苷酸
[0405]
具有预定序列的双链多核苷酸通过本发明的方法通过将预定的核苷酸合并预先存在的多核苷酸中来合成,所述多核苷酸在本文中称为支架多核苷酸,其可附接于或能够附接于表面,如本文所述。如本文更详细描述的,支架多核苷酸形成支持结构以容纳新合成的多核苷酸,并且如从本文的描述中显而易见的,其不包括如常规合成方法中那样复制的预先存在的模板链。如果支架多核苷酸附接于表面,则支架多核苷酸可称为锚多核苷酸。本文更详细地描述了用于将支架多核苷酸附接到表面以形成锚多核苷酸的表面附接化学。
[0406]
在一个实施方案中,支架多核苷酸包括与互补支持链杂交的合成链。合成链包含引物链部分(例如参见图1至5)。可以提供与互补支持链杂交的合成链。或者,可以分开提供支持链和合成链,然后使其杂交。
[0407]
可以提供支架多核苷酸,其中每个支持和合成链在相邻末端不连接。支架多核苷酸可以在支架多核苷酸的两端提供有支持和合成链,它们在相邻末端连接,例如通过发夹环。支架多核苷酸可以在支架多核苷酸或任何其它合适的接头的一端提供有支持和合成链,它们在相邻末端连接,例如通过发夹环。
[0408]
如本文中更详细描述(参见图11),具有或不具有发夹的支架多核苷酸可固定到固体载体或表面。
[0409]
术语“发夹”或“发夹环”通常用于当前技术领域。术语“发夹环”通常也称为“茎环”。这些术语是指多核苷酸中的二级结构区域,其包括未配对核碱基的环,当多核苷酸分
子的一条链由于分子内碱基配对而与相同链的另一部分杂交时形成。因此发夹可以类似于u形结构。这种结构的实例如图11所示。
[0410]
在本文所述的某些方法中,通过连接酶的作用将预定序列的第一核苷酸合并到支架多核苷酸中,从而开始新的合成。因此,如本文进一步所述,预定序列的第一核苷酸与支架多核苷酸的支持链的末端核苷酸连接。预定义序列的第一核苷酸由多核苷酸连接分子提供,该多核苷酸连接分子包括支持链、辅助链和互补的连接端。提供预定序列的第一核苷酸作为互补连接端的支持链的末端核苷酸。
[0411]
辅助链在互补连接端的末端核苷酸是不可连接的核苷酸,并且通常提供为缺少磷酸基团。这防止了辅助链的末端核苷酸与支架多核苷酸的引物链部分的末端核苷酸连接,并且在连接后在辅助链和引物链部分之间产生了单链断裂位点。单链断裂的产生和维持可以通过其它方式实现。例如,辅助链部分的末端核苷酸可以具有合适的封闭基团,其阻止与引物链部分的连接。
[0412]
在本文所述的某些方法中,通过聚合酶或转移酶的作用将预定序列的第二核苷酸合并到支架多核苷酸中。因此,聚合酶或转移酶将起作用以延伸引物链部分的末端核苷酸。
[0413]
本文进一步提供了示例性方法的一般方法方案的更多细节。
[0414]
核苷酸和通用核苷酸
[0415]
可以通过本文描述的任何方法合并合成多核苷酸中的核苷酸可以是核苷酸、核苷酸类似物和修饰的核苷酸。
[0416]
核苷酸可包括天然核碱基或非天然核碱基。核苷酸可含有天然核碱基、糖和磷酸基团。天然核碱基包括腺苷(a)、胸腺嘧啶(t)、尿嘧啶(u)、鸟嘌呤(g)和胞嘧啶(c)。可进一步修饰核苷酸的一种组分。
[0417]
核苷酸类似物是在碱基、糖或磷酸盐或其组合中在结构上被修饰并且仍然是聚合酶可接受作为合并寡核苷酸链的底物的核苷酸。
[0418]
非天然核碱基可为在一定程度上将键结,例如氢键结到目标多核苷酸中的所有核碱基的核碱基。非天然核碱基优选地是在一定程度上将键结,例如氢键结到包括核苷腺苷(a)、胸腺嘧啶(t)、尿嘧啶(u)、鸟嘌呤(g)和胞嘧啶(c)的核苷酸的核碱基。
[0419]
非天然核苷酸可为肽核酸(pna)、锁核酸(lna)和解锁核酸(una)、桥接核酸(bna)或吗啉代、硫代磷酸酯或甲基膦酸酯。
[0420]
非天然核苷酸可以包括经修饰的糖和/或经修饰的核碱基。经修饰的糖包含但不限于2'-o-甲基核糖。经修饰的核碱基包含但不限于甲基化的核碱基。核碱基的甲基化是表观遗传修饰的公认形式,其具有改变基因和其它元件(如微rna)的表达的能力。核碱基的甲基化发生在离散的基因座处,所述基因座主要是由cpg基序组成的二核苷酸,但也可以在chh基序(其中h是a、c或t)处发生。通常,在甲基化过程中,将甲基加到胞嘧啶碱基的第五个碳上以产生甲基胞嘧啶。因此,修饰的核碱基包含但不限于5-甲基胞嘧啶。
[0421]
预定序列的核苷酸可以与配偶体核苷酸相对合并以形成核苷酸对。配偶体核苷酸可为互补核苷酸。互补核苷酸为在一定程度上能够键结,例如氢键结到预定义序列的核苷酸的核苷酸。
[0422]
通常,将预定序列的核苷酸合并多核苷酸中与天然互补的配偶体核碱基相对。因此,可以合并腺苷,与胸腺嘧啶相对,反之亦然。可以合并鸟嘌呤,与胞嘧啶相对,反之亦然。
可替代地,可以合并预定序列的核苷酸,与其在一定程度上将键合例如氢键合的配偶体核碱基相对。
[0423]
或者,配偶体核苷酸可为非互补核苷酸。非互补核苷酸是这样的核苷酸,其不能够键结,例如氢键结到预定义序列的核苷酸。因此,预定序列的核苷酸可以与配偶体核苷酸相对合并以形成错配,条件是合成的多核苷酸总体上是双链的,并且其中第一链通过杂交与第二链连接。
[0424]
术语“相对”应理解为涉及所述术语在核酸生物化学领域中的正常使用,并且具体地涉及常规的watson-crick碱基配对。因此,序列5'-acga-3'的第一核酸分子可与序列5'-tcgt-3'的第二核酸分子形成双链体,其中第一分子的g将位于与第二分子的c相对并与之形成氢键。序列5'-atga-3'的第一核酸分子可以与序列5'-tcgt-3'的第二核酸分子形成双链体,其中第一分子的t与第二分子的g错配,但仍然位于与其相对,并将作为配偶体核苷酸。所述原理适用于本文公开的任何核苷酸配偶体对关系,包含包括通用核苷酸的配偶体对。
[0425]
在本文所述的所有方法中,支持链中的位置和合成链中的相对位置被指定为位置编号“n”。该位置是指支架多核苷酸的支持链中核苷酸的位置,该核苷酸在任何给定的合成循环中与合成链中被预定序列的第二或其他核苷酸占据或将被其占据的核苷酸位置相对(在该循环中或在随后循环的合并步骤中将其添加到引物链部分的末端时)。位置“n”还指在连接步骤之前多核苷酸连接分子在支持链中的位置,该位置是与多核苷酸连接分子支架多核苷酸连接以及通过聚合酶或转移酶的作用合并预定序列的第二或其他核苷酸后将与预定序列的第二或其他核苷酸相对的核苷酸位置。
[0426]
支持链中的位置和合成链中的相对位置都可以称为位置n。
[0427]
参考图1至图5及其关于本发明的示例性合成方法版本及其在此更详细描述的变型的描述,提供了关于位置“n”的定义的更多细节。
[0428]
核苷酸和核苷酸类似物可以优选作为核苷三磷酸提供。因此,在本发明的任何方法中,为了合成dna多核苷酸,可以从2
′-
脱氧核糖核苷-5
′-
o-三磷酸酯(dntp)中合并核苷酸,例如通过dna聚合酶的作用或通过具有脱氧核苷酸末端转移酶活性的酶的作用。在本发明的任何方法中,为了合成rna多核苷酸,可以将核苷酸合并核糖核苷5
′-
o-三磷酸酯(ntp),例如通过rna聚合酶的作用或例如通过具有核苷酸末端转移酶活性的酶的作用。三磷酸可以被四磷酸或五磷酸(一般低聚磷酸)取代。这些低聚磷酸可以被其它烷基或酰基取代:
[0429]
[0430]
本发明的方法可以使用通用核苷酸。通用核苷酸可以用作支架分子的支持链的组分,以促进新合并的核苷酸被正确地在合成的每一个循环期间与其期望的配偶体核苷酸配对。如果需要,通用核苷酸也可以作为预定核苷酸序列的组分合并合成链中。
[0431]
通用核苷酸是其中核碱基将在某种程度上与预定义序列的任何核苷酸的核苷碱基键结,例如氢键结的核苷酸。通用核苷酸优选地是将在某种程度上与包括核苷腺苷(a)、胸腺嘧啶(t)、尿嘧啶(u)、鸟嘌呤(g)和胞嘧啶(c)的核苷酸键结,例如氢键结的核苷酸。与其它核苷酸相比,通用核苷酸可以更强地与一些核苷酸键结。例如,包括核苷2'-脱氧肌苷的通用核苷酸(i)将示出i-c>i-a>i-g约=i-t的优先配对顺序。
[0432]
可能的通用核苷酸的实例是肌苷或硝基吲哚。通用核苷酸优选地包括以下核碱基中的一个:次黄嘌呤、4-硝基吲哚、5-硝基吲哚、6-硝基吲哚、3-硝基吡咯、硝基咪唑、4-硝基吡唑、4-硝基苯并咪唑、5-硝基吲唑、4-氨基苯并咪唑或苯基(c6-芳环)。通用核苷酸更优选地包括以下核苷之一:2'-脱氧肌苷、肌苷、7-脱氮-2'-脱氧肌苷、7-脱氮-肌苷、2-氮杂-脱氧肌苷、2-氮杂-肌苷、4-硝基吲哚2'-脱氧核糖核苷、4-硝基吲哚核糖核苷、5-硝基吲哚2'脱氧核糖核苷、5-硝基吲哚核糖核苷、6-硝基吲哚2'脱氧核糖核苷、6-硝基吲哚核糖核苷、3-硝基吡咯2'脱氧核糖核苷、3-硝基吡咯核糖核苷、次黄嘌呤的非环糖类似物、硝基咪唑2'脱氧核糖核苷、硝基咪唑核糖核苷、4-硝基吡唑2'脱氧核糖核苷、4-硝基吡唑核糖核苷、4-硝基苯并咪唑2'脱氧核糖核苷、4-硝基苯并咪唑核糖核苷、5-硝基吲唑2'脱氧核糖核苷、5-硝基吲唑核糖核苷、4-氨基苯并咪唑2'脱氧核糖核苷、4-氨基苯并咪唑核糖核苷、苯基c-核糖核苷或苯基c-2'-脱氧核糖基核苷。
[0433]
通用碱基的一些实例如下所示:
[0434][0435]
还可以使用合并可裂解碱基的通用核苷酸,包含光可裂解碱基和酶可裂解碱基,所述碱基的一些实例如下所示。
[0436]
光可裂解碱基:
[0437][0438]
可通过核酸内切酶iii裂解的碱基类似物:
[0439][0440]
可通过甲酰胺基嘧啶dna糖基化酶(fpg)裂解的碱基类似物:
[0441][0442]
可通过8-氧代鸟嘌呤dna糖基化酶(hogg1)裂解的碱基类似物:
[0443][0444]
可通过hneil1裂解的碱基类似物:
[0445][0446]
可通过胸腺嘧啶dna糖基化酶(tdg)裂解的碱基类似物:
[0447][0448]
可通过人类烷基腺嘌呤dna糖基化酶(haag)裂解的碱基类似物:
[0449][0450]
可通过尿嘧啶dna糖基化酶裂解的碱基:
[0451][0452]
可通过人类单链选择性单功能的尿嘧啶-dna糖基化酶(smug1)裂解的碱基:
[0453][0454]
可通过5-甲基胞嘧啶dna糖基化酶(ros1)裂解的碱基:
[0455][0456]
(参见s.s.david,s.d.williams chemical reviews 1998,98,1221-1262和m.i.ponferrada-mar
í
n,t.rold
á
n-arjona,r.r.ariza'nucleic acids res 2009,37,4264-4274)。
[0457]
在涉及支架多核苷酸的任何方法中,通用核苷酸最优选包括2'-脱氧肌苷。
[0458]
可以使用本文描述的任何合成方法合并的表观遗传碱基的实例包括以下:
[0459][0460]
可以使用本文描述的任何合成方法合并的修饰碱基的实例包括以下:
[0461][0462]
可使用本文所述的任何合成方法合并的卤代碱基的实例包括以下:
[0463][0464]
其中r1=f、cl、br、i、烷基、芳基、荧光标记、氨基炔丙基、氨基烯丙基。
[0465]
其可以使用本文描述的任何合成方法合并的可以用于例如附接/接头化学的氨基修饰的碱基的实例包括以下:
[0466][0467]
其中碱基=a、t、g或c,具有炔烃或烯烃接头。
[0468]
其可以使用本文描述的任何合成方法合并的可以用于例如点击化学的修饰的碱基的实例包括以下:
[0469][0470]
可以使用本文描述的任何合成方法合并的生物素修饰的碱基的实例包括以下:
[0471]
[0472]
其中碱基=a、t、g或c,具有炔烃或烯烃接头。
[0473]
可以使用本文所述的任何合成方法合并的带有荧光团和猝灭剂的碱基的实例包括以下:
[0474][0475]
核苷酸合并酶
[0476]
可获得的酶能够通过添加核苷酸来延伸双链多核苷酸分子的单链多核苷酸部分和/或能够延伸平端双链多核苷酸分子的一条链的酶。这包含具有模板独立性酶活性的酶,如模板独立性聚合酶或模板独立性转移酶活性。
[0477]
因此,在本文所述的任何方法中,所述酶用于将预定序列的第二核苷酸和/或预定序列的另一核苷酸添加至支架多核苷酸的合成链的引物链部分的末端,其具有独立于模板的酶活性,例如独立于模板的聚合酶或独立于模板的转移酶活性。
[0478]
使用本文所述的方法,可以采用任何合适的酶来添加预定的核苷酸。因此,在本文中所定义和描述的所有方法中,参考聚合酶或转移酶的使用,聚合酶或转移酶可以用能够在本发明的方法的情形下与聚合酶或转移酶执行相同功能的另一种酶取代。
[0479]
聚合酶可用于本文所描述的方法中。可以基于它们合并修饰的核苷酸的能力来选择聚合酶,特别是具有连接的可逆终止子基团的核苷酸,如本文所述。在本文所述的示例性方法中,作用于dna的所有聚合酶必须不具有3'至5'核酸外切酶活性。聚合酶可具有链置换活性。
[0480]
因此,优选地,聚合酶是修饰的聚合酶,其与未修饰的聚合酶相比具有增强的合并包含可逆终止子基团的核苷酸的能力。聚合酶更优选地是来自嗜热球菌(thermococcus)物种9
°
n,优选地物种9
°
n-7的天然dna聚合酶的基因工程变体。修饰的聚合酶的实例是可从new england biolabs获得的终止子ix dna聚合酶和终止子x dna聚合酶。该酶具有增强的合并3'-o-修饰的dntp的能力。
[0481]
可用于在本发明的任何方法中合并可逆终止子dntp的其他聚合酶的实例是deep vent(exo-)、vent(exo-)、9
°
n dna聚合酶、终止子dna聚合酶、therminator ix dna聚合酶、终止子x dna聚合酶、klenow片段(exo-)、bst dna聚合酶、bsu dna聚合酶、sulfolobus dna聚合酶i和taq聚合酶。
[0482]
可用于本发明任何方法中的可逆终止子ntp合并的其他聚合酶的实例是t3 rna聚合酶、t7 rna聚合酶、sp6 rna聚合酶、pol lambda、pol micro或φ29dna聚合酶。
[0483]
对于包括dna的这种多核苷酸合成分子的延伸,可以使用dna聚合酶。可以使用任何合适的dna聚合酶。
[0484]
dna聚合酶可以是例如bst dna聚合酶全长、bst dna聚合酶大片段、bsu dna聚合
酶大片段、大肠杆菌dna聚合酶dna pol i大(克列诺(klenow))片段、m-mulv逆转录酶、phi29 dna聚合酶、硫化叶菌dna聚合酶iv、taq dna聚合酶、t4 dna聚合酶、t7 dna聚合酶和具有逆转录酶活性的酶,例如m-mulv逆转录酶。
[0485]
dna聚合酶可能缺乏3'到5'核酸外切酶活性。可以使用任何这种合适的聚合酶。这种dna聚合酶可以是例如bst dna聚合酶全长、bst dna聚合酶大片段、bsu dna聚合酶大片段、dna pol i大(克列诺)片段(3
’→5’
体外-)、m-mulv逆转录酶、硫化叶菌dna聚合酶iv、taq dna聚合酶。
[0486]
dna聚合酶可具有链置换活性。可以使用任何这种合适的聚合酶。这种dna聚合酶可以是例如bst dna聚合酶大片段、bsu dna聚合酶大片段、dna pol i大(克列诺)片段(3
’→5’
体外-)、m-mulv逆转录酶、phi29 dna聚合酶。
[0487]
dna聚合酶可能缺乏3'到5'核酸外切酶活性,并且可以具有链置换活性。可以使用任何这种合适的聚合酶。这种dna聚合酶可以是例如bst dna聚合酶大片段、bsu dna聚合酶大片段、大肠杆菌dna聚合酶dna pol i大(克列诺)片段、m-mulv逆转录酶。
[0488]
dna聚合酶可能缺乏5'到3'核酸外切酶活性。可以使用任何这种合适的聚合酶。这种dna聚合酶可以是例如,bst dna聚合酶大片段、bsu dna聚合酶大片段、dna pol i大(克列诺)片段、dna pol i大(克列诺)片段(3
’→5’
体外-)、m-mulv逆转录酶、phi29 dna聚合酶、硫化叶菌dna聚合酶iv、t4 dna聚合酶、t7 dna聚合酶。
[0489]
dna聚合酶可能缺乏3'到5'和5'到3'核酸外切酶活性两者,并且可以具有链置换活性。可以使用任何这种合适的聚合酶。这种dna聚合酶可以是例如bst dna聚合酶大片段、bsu dna聚合酶大片段、dna pol i大(克列诺)片段(3
’→5’
体外-)、m-mulv逆转录酶。
[0490]
dna聚合酶也可以是经过基因工程化的变体。例如,dna聚合酶可以是来自嗜热球菌物种9
°
n,例如物种9
°
n-7的天然dna聚合酶的基因工程变体。经过修饰的聚合酶的一个这种实例是可从新英格兰生物实验室(new england biolabs)获得的终止子ix dna聚合酶或终止子x dna聚合酶。其它经过工程化或变体dna聚合酶包含deep vent(exo-)、vent(exo-)、9
°
n dna聚合酶、终止子dna聚合酶、克列诺片段(exo-)、bst dna聚合酶、bsu dna聚合酶、硫化叶菌dna聚合酶i和taq聚合酶。
[0491]
为了延伸这种包含rna的多核苷酸合成分子,可以使用任何合适的酶。例如,可以使用rna聚合酶。可以使用任何合适的rna聚合酶。
[0492]
rna聚合酶可为t3 rna聚合酶、t7 rna聚合酶、sp6 rna聚合酶、大肠杆菌rna聚合酶全酶。
[0493]
酶可以具有末端转移酶活性,例如酶可以是末端核苷酸转移酶或末端脱氧核苷酸转移酶,并且其中多核苷酸合成分子经延伸以形成包括dna或rna,优选dna的多核苷酸分子。这些酶中的任何一种酶可以用于本发明的方法中,其中需要延伸多核苷酸合成分子。
[0494]
一种此类酶是末端核苷酸转移酶,如末端脱氧核苷酸转移酶(tdt)(参见例如,motea等人,2010;minhazud-dean,syst.synth.biol.,2008,2(3-4),67
–
73)。tdt能够催化来自核苷三磷酸基质(ntp或dntp)的核苷酸分子(核苷单磷酸)向多核苷酸合成分子的加成。tdt能够催化天然和非天然核苷酸的添加。其还能够催化核苷酸类似物的加成(motea等人,2010)。还可以使用polλ和polμ酶(ramadan k,等人,j.mol.biol.,2004,339(2),395-404),如可以使用φ29dna聚合酶。
[0495]
本领域已经广泛讨论了通过末端转移酶(例如,末端脱氧核苷酸转移酶;tdt)的作用在不存在模板的情况下延伸单链多核苷酸分子dna和rna两者以产生人工合成的单链多核苷酸分子的技术。这些技术在例如专利申请公开wo2016/034807、wo 2016/128731、wo2016/139477和wo2017/009663,以及us2014/0363852、us2016/0046973、us2016/0108382和us2016/0168611中公开。这些文献描述了通过tdt的作用产生人工合成的单链多核苷酸分子的单链多核苷酸合成分子的受控延伸。描述了使用这种酶通过天然和非天然/人工核苷酸的延伸,如通过经过修饰的核苷酸的延伸,例如合并封闭基团的核苷酸的延伸。这些文献中公开的任何末端转移酶和其任何酶片段、衍生物、类似物或功能等同物均可以应用于本发明的方法中,条件是末端转移酶功能被保存在所述酶中。
[0496]
定向进化技术、常规筛选、合理或半合理工程化/诱变方法或任何其它合适的方法可以用于改变任何此类酶以提供和/或优化所需功能。可以使用能够不使用模板而延伸单链多核苷酸分子部分的任何其他酶,例如包含dna或rna的分子,或具有核苷酸的平末端分子的一条链。
[0497]
因此,在本文定义的任何方法中,包括dna的单链多核苷酸合成分子或包括dna的平端双链多核苷酸可以通过酶延伸,所述酶具有模板独立性酶活性,如模板独立性聚合酶或转移酶活性。酶可以具有核苷酸转移酶活性,例如脱氧核苷酸转移酶,例如末端脱氧核苷酸转移酶(tdt)或其酶片段、衍生物、类似物或功能等效物。通过这种酶的作用延伸的多核苷酸合成分子包括dna。
[0498]
在本文定义的任何方法中,包含rna的多核苷酸合成分子的单链部分或包含rna的平末端双链多核苷酸可被具有核苷酸转移酶(例如包括tdt)或酶片段、其衍生物、类似物或功能等同物延伸。通过这种酶的作用延伸的多核苷酸合成分子可以包括rna。对于包括rna的单链多核苷酸合成分子或包括rna的多核苷酸合成分子的单链部分的合成,可以使用任何合适的核苷酸转移酶。核苷酸转移酶,例如聚(u)聚合酶和聚(a)聚合酶(例如来自大肠杆菌)能够将核苷酸单磷酸单元不依赖模板地添加道多核苷酸合成分子。这些酶中的任何一种酶以及其任何酶片段、衍生物、类似物或功能等同物可以应用于本发明的方法中,条件是核苷酸转移酶功能被保存在所述酶中。定向进化技术、常规筛选、合理或半合理工程化/诱变方法或任何其它合适的方法可以用于改变任何此类酶以提供和/或优化所需功能。
[0499]
可逆终止子基团
[0500]
在本文定义和描述的本发明的任何合成方法中,优选将通过聚合酶或转移酶的作用合并到合成链中的核苷酸作为包含一个或多个可逆性封闭基团的核苷酸合并,所述核苷酸也被称为本文所述的可逆终止基团。
[0501]
这些基团的作用是阻止酶在给定的合成循环中进一步延伸作用,使得只有预定序列的核苷酸可以可控制地用于延伸合成链,因此防止了非特异性核苷酸合并。实现所述效果的任何功能可以用于本文限定和描述的任何方法中。与核苷酸连接的可逆封闭基团/可逆终止子基团和解封闭步骤是实现所述效果的优选方法。然而,这种效果可以通过适当的替代方式来实现。
[0502]
任何合适的可逆性保护基团都可以连接到核苷酸上,以防止在给定的循环中将核苷酸合并到合成链中后酶进一步延伸,并限制每一步合并到合成链中的一个核苷酸。在本发明的任何方法中,可逆封闭基团优选是可逆终止子基团,其起到防止聚合酶进一步延伸
的作用。以下提供可逆终止子的实例。
[0503]
炔丙基可逆终止子:
[0504][0505]
烯丙基可逆终止子:
[0506][0507]
环辛烯可逆终止子:
[0508][0509]
氰乙基可逆终止子:
[0510][0511]
硝基苄基可逆终止子:
[0512]
[0513]
二硫化物可逆终止子:
[0514][0515]
叠氮甲基可逆终止子:
[0516][0517]
氨基烷氧基可逆终止子:
[0518][0519]
具有与碱基连接的庞大基团的核苷三磷酸可以作为3'-羟基上的可逆终止子基团的替代物并且可以阻止进一步合并。所述基团可以通过tcep或dtt脱保护,产生天然核苷酸。
[0520][0521]
为了根据本发明的任何方法合成dna多核苷酸,优选的修饰核苷是3'-o-修饰的-2'-脱氧核糖核苷-5'-o-三磷酸。为了根据本发明的任何方法合成rna多核苷酸,优选的修饰核苷是3'-o-修饰的-核糖核苷-5'-o-三磷酸。
核苷酸(与pd0结合)和3'-o-叠氮甲基-核苷酸去除可逆终止子基团。
[0528]
以下提供了脱保护试剂的实例。
[0529]
炔丙基可逆终止子:
[0530]
用pd催化剂-na2pdcl4、pdcl2处理。
[0531]
可以使用配体,例如:三苯基膦-3,3',3
”-
三磺酸三钠盐。
[0532]
烯丙基可逆终止子:
[0533]
用pd催化剂-na2pdcl4、pdcl2处理。
[0534]
可以使用配体,例如:三苯基膦-3,3',3
”-
三磺酸三钠盐。
[0535]
叠氮甲基可逆终止子:
[0536]
通过硫醇(巯基乙醇或二硫苏糖醇)或三(2-羧乙基)膦-tcep处理。
[0537]
氰乙基可逆终止子:
[0538]
通过氟化物-氟化铵、四丁基氯化铵(tbaf)处理。
[0539]
硝基苄基可逆终止子:
[0540]
暴露在uv光下。
[0541]
二硫化物可逆终止子:
[0542]
通过硫醇(巯基乙醇或二硫苏糖醇)或三(2-羧乙基)膦-tcep处理。
[0543]
氨基烷氧基可逆终止子:
[0544]
用亚硝酸盐(no
2-、hno2)ph=5.5处理
[0545]
可以通过在合并步骤之后立即进行的步骤除去可逆的保护基团(例如,可逆的终止剂基团),条件是从合并步骤中移除不需要的试剂以防止在移除可逆的终止子基团之后进一步合并。
[0546]
多核苷酸连接分子
[0547]
在需要存在支架多核苷酸和连接步骤的方法中,多核苷酸连接分子的构型和结构的选择也将取决于所采用的特定方法。多核苷酸连接分子通常包含如本文所述的支持链和如本文所述的辅助链。多核苷酸连接分子在分子的一端包含互补的连接端。多核苷酸连接分子的互补连接端将被连接至支架多核苷酸的末端。
[0548]
多核苷酸连接分子的互补连接端在辅助链中具有不可连接的末端核苷酸,通常是非磷酸化的末端核苷酸。这防止了合成链的辅助链部分与合成链的引物链部分的连接,因此在连接后在合成链中产生了单链断裂。可以使用用于防止合成链中连接的替代方法。例如,封闭部分可以与辅助链中的末端核苷酸连接。此外,可以在裂解之前从支架分子中移除辅助链,例如通过变性移除,如在本文中进一步描述的。多核苷酸连接分子的互补连接端在支持链中与辅助链中不可连接的末端核苷酸相邻地具有可连接的末端核苷酸。支持链的可连接的末端核苷酸是通过连接酶的作用而被合并到支架分子中的预定序列的第一核苷酸。多核苷酸连接分子的互补连接端在支持链中也具有通用核苷酸。相对于支持链的可连接的末端核苷酸,支持链中通用核苷酸的确切定位将取决于所采用的具体反应化学,这将从具体方法版本及其变体的描述中显而易见。
[0549]
通过参考本文描述的示例性方法及其在附图中的描绘,可以容易地确定多核苷酸连接分子的适当结构。
[0550]
连接
[0551]
在涉及连接步骤的本发明方法中,可以使用任何合适的方法实现连接。优选地,连接步骤将通过连接酶进行。连接酶可以是修饰的连接酶。连接酶可以是t3 dna连接酶或t4 dna连接酶。连接酶可以是平端ta连接酶(blunt ta ligase)。例如,可从新英格兰生物实验室(neb)获得平端ta连接酶。这是t4 dna连接酶、连接增强剂和优化的反应缓冲液的即用型预混液,该缓冲液经特殊配制以改善平末端底物的连接和转化。用于连接(连结)单链和双链多核苷酸的分子、酶、化学品和方法是本领域技术人员公知的。
[0552]
支架多核苷酸的裂解
[0553]
在需要存在支架多核苷酸和裂解步骤的方法中,进行裂解步骤的试剂的选择将取决于所采用的特定方法。在支持链中,裂解位点由通用核苷酸的特定位置限定。因此,所需裂解位点的构型和适当裂解试剂的选择将取决于该方法中采用的特定化学,通过参考本文所述的示例性方法将很容易明白。
[0554]
识别修饰的碱基的dna裂解酶的一些实例示出在下表中。
[0555][0556]
合成链
[0557]
在本文描述的合成多核苷酸或寡核苷酸的方法中,包括但不限于图1至图5以及在本文中进一步描述的本发明的合成方法版本1和2及其变体,支架多核苷酸具有合成链。合成链包含引物链部分。在合成循环期间,通过延伸引物链部分,将预定序列的每个新的第二核苷酸合并合成链,将预定序列的第一核苷酸合并支持链。诸如聚合酶或具有末端转移酶活性的酶的酶可用于催化每个新的第二核苷酸的合并/添加。预定序列的每个新结合的第二核苷酸将充当引物链部分的末端核苷酸,用于在下一个结合步骤中引发结合。因此,在任
何给定的合成循环中,合成链的引物链部分将包含足够的多核苷酸序列以允许通过适当的酶引发。在本文进一步描述的某些实施方案中,在给定的合成循环中,将预定序列的第二核苷酸合并到合成链中,然后将一个或多个其他核苷酸合并到合成链中。在这样的实施方案中,预定序列的第二核苷酸和其他核苷酸包含可逆终止子基团,并且该方法另外包括在合并下一核苷酸之后和在合并下一核苷酸之前从核苷酸中除去可逆终止子基团的步骤。
[0558]
核苷酸的术语“合并”、“延伸”和“添加”在本文中具有相同的含义。
[0559]
辅助链
[0560]
可以在多核苷酸连接分子中提供辅助链,以在连接步骤中促进多核苷酸连接分子与支架多核苷酸的连接。辅助链还可在裂解步骤中促进裂解酶的结合。可以省略辅助链,条件是提供替代方式以确保在裂解步骤中裂解酶的结合并且如果需要的话确保在连接步骤中的连接。在本发明的优选方法中,合成链具有辅助链。
[0561]
对辅助链的长度、序列和结构的参数没有特殊要求,条件是如有需要时辅助链适合于促进连接酶和裂解酶的结合。
[0562]
辅助链可包含核苷酸、核苷酸类似物/衍生物和/或非核苷酸。
[0563]
优选地,应所述避免辅助链的序列区域内与支持链的错配,应避免富含gc和at的区域,此外应避免二级结构的区域,例如发夹或凸起。
[0564]
辅助链的长度可以是10个碱基或更多。任选地,辅助链的长度可以是15个碱基或更多,优选30个碱基或更多。然而,辅助链的长度可以变化,条件是辅助链能够促进裂解和/或连接。
[0565]
辅助链必须与支持链的相应区域杂交。如果辅助链可以促进连接步骤中连接酶的结合和/或裂解步骤中裂解酶的结合,则整个辅助链与支持链的相应区域杂交不是必需的。因此,可以容许辅助链和支持链的相应区域之间的错配。辅助链可以比支持链的相应区域长。支持链可以在远离引物链的方向上延伸超出与辅助链相对应的区域。辅助链可以例如通过发夹连接到支持链的相应区域。
[0566]
辅助链可以与支持链杂交,使得缺口位点处的辅助链的末端核苷酸占据合成链中相对于缺口位点处的引物链部分的末端核苷酸的下一顺序核苷酸位置。因此,在所述构型中,辅助链和引物链之间没有核苷酸位置切口。然而,由于存在单链断裂或缺口,辅助链和引物链将是物理分离的。
[0567]
与通用核苷酸配对的辅助链中的核苷酸可以是任何合适的核苷酸。优选地,应所述避免可能扭曲分子螺旋结构的配对。优选地,胞嘧啶充当通用核苷酸的配偶体。在特别优选的实施方案中,通用核苷酸是肌苷或其类似物、变体或衍生物,并且辅助链中通用核苷酸的配偶体核苷酸是胞嘧啶。
[0568]
移除辅助链
[0569]
在本文描述的本发明的任何合成方法中,在合并预定序列的第二核苷酸的步骤之前,可以将多核苷酸连接分子提供的辅助链从支架多核苷酸中除去。
[0570]
合成链的辅助链部分可以通过任何合适的方法从支架多核苷酸中移除,包括但不限于:(i)将支架多核苷酸加热至约80℃至约95℃的温度并将辅助链部分与支架多核苷酸分离,(ii)用尿素溶液(例如8m尿素)处理支架多核苷酸并将辅助链部分与支架多核苷酸分离,(iii)用甲酰胺或甲酰胺溶液(例如100%甲酰胺)处理支架多核苷酸并将辅助链部分与
支架多核苷酸分离,或(iv)使支架多核苷酸与单链多核苷酸分子接触,所述单链多核苷酸分子包含与辅助链部分的序列互补的核苷酸序列区域,从而竞争性地抑制辅助链部分与支架多核苷酸的杂交。
[0571]
在将双链多核苷酸连接分子连接至裂解的支架多核苷酸的步骤之后且在裂解支架多核苷酸的步骤之前,将辅助链部分从支架多核苷酸中移除的方法中,裂解步骤将包括在不存在辅助链提供的双链区的情况下裂解支持链。可以选择任何合适的酶来进行这样的裂解步骤,例如选自本文公开的任何合适的酶。
[0572]
引物链
[0573]
引物链部分应适合于允许酶(例如聚合酶或具有末端转移酶活性的酶)引发合成,即催化在引物链部分末端添加新核苷酸。
[0574]
引物链可以包括可以用于引发新的多核苷酸合成的序列区域(例如如图1到5中的每一个中描绘的结构中的虚线所示)。引物链可由可作用于引发新多核苷酸合成的序列区域组成,因此引物链的全部可为可作用于引发如本文所述的新多核苷酸合成的序列。
[0575]
对引物链的长度、序列和结构的参数没有特殊要求,条件是引物链适合引发新的多核苷酸合成。
[0576]
引物链可包括核苷酸、核苷酸类似物/衍生物和/或非核苷酸。
[0577]
技术人员能够容易地构建能够引发新的多核苷酸合成的引物链。因此,在可以起到引发新的多核苷酸的作用的引物链的序列区域内,应所述避免与支持链的错配,应避免富含gc和at的区域,以及此外应避免二级结构的区域,如发夹或凸起。
[0578]
本领域技术人员可根据需要和所用的聚合酶选择可用于引发新的多核苷酸合成的引导链的序列区域的长度。所述区域的长度可以是7个碱基或更多、8个碱基或更多、9个碱基或更多个或10个碱基或更多。任选地,所述区域的长度为15个碱基或更多,优选30个碱基或更多。
[0579]
引物链必须与支持链的相应区域杂交。如果引物链能够引发新的多核苷酸合成,则整个引物链与支持链的相应区域杂交不是必需的。因此,可以在一定程度上容许引物链和支持链的相应区域之间的错配。优选地,可用于引发新多核苷酸合成的引物链序列区域应包括与支持链中相应核碱基互补的核碱基。
[0580]
引物链可以例如通过发夹连接到支持链的相应区域。
[0581]
支持链
[0582]
在包含但不限于如上文所描述的本发明的合成方法版本1到2的本发明的方法中,支架多核苷酸具备支持链。支持链与合成链杂交。对支持链的长度、序列和结构的参数没有特殊要求,只要该支持链与本文所述的引物链部分以及所述合成链的辅助链部分(如果存在)相容即可,如上所述。
[0583]
合成多核苷酸
[0584]
具有根据本文所述方法合成的预定序列的多核苷酸是双链的。合成的多核苷酸总体上是双链的,并且其中第一链通过杂交与第二链连接。只要整个第一链通过杂交与第二链连接,可以容许错配和非杂交区域。
[0585]
杂交可以通过中等严格或严格杂交条件来定义。中等严格的杂交条件是使用含有5x氯化钠/柠檬酸钠(ssc)、0.5%sds、1.0mm edta(ph 8.0)、约50%甲酰胺的杂交缓冲液、
6xssc且杂交温度为55℃的预洗涤溶液(或其他类似的杂交溶液,例如含有约50%甲酰胺的溶液,杂交温度为42℃),并且洗涤条件为60℃,在0.5xssc、0.1%sds中进行。严格的杂交条件在45℃下于6xssc中杂交,然后在68℃下于0.1xssc,0.2%sds中进行一次或多次洗涤。
[0586]
具有根据本文所述方法合成的预定义序列的双链多核苷酸可以保留为双链多核苷酸。或者,可以分离双链多核苷酸的两条链以提供具有预定义序列的单链多核苷酸。允许双链多核苷酸的两条链分离的条件(熔融)是本领域公知的(例如,sambrook等人,2001,molecular cloning:a laboratory manual,3rd edition,cold spring harbour laboratory press;和current protocols in molecular biology,greene publishing and wiley-lnterscience,new york(1995))。
[0587]
具有根据本文所述方法合成的预定义序列的双链多核苷酸可以在合成后扩增。可以扩增双链多核苷酸的任何区域。双链多核苷酸的全部或任何区域可以与支架多核苷酸的全部或任何区域一起扩增。允许双链多核苷酸扩增的条件是本领域公知的(例如,sambrook等人,2001,molecular cloning:a laboratory manual,3rd edition,cold spring harbour laboratory press;和current protocols in molecular biology,greene publishing and wiley-lnterscience,new york(1995))。因此,本文所述的任何合成方法可以进一步包括扩增步骤,其中如上所述扩增具有预定义序列的双链多核苷酸或其任何区域。可以通过任何合适的方法执行扩增,如聚合酶链反应(pcr)、聚合酶螺旋反应(psr)、环介导的等温扩增(lamp)、基于核酸序列的扩增(nasba)、自主序列复制(3sr)、滚环扩增(rca)、链置换扩增(sda)、多重置换扩增(mda)、连接酶链反应(lcr)、解旋酶依赖性扩增(hda)、网状分支扩增方法(ram)等。优选地,通过聚合酶链反应(pcr)执行扩增。
[0588]
具有根据本文所述方法合成的预定义序列的双链或单链多核苷酸可以是任何长度。例如,多核苷酸的长度可以是至少10、至少50、至少100、至少150、至少200、至少250、至少300、至少350、至少400、至少450或至少500核苷酸或核苷酸对。例如,长度上,多核苷酸可以是约10至约100核苷酸或核苷酸对,约10至约200核苷酸或核苷酸对,约10至约300核苷酸或核苷酸对,约10至约400核苷酸或核苷酸对和约10至约500核苷酸或核苷酸对。多核苷酸可以是多达约1000个或更多核苷酸或核苷酸对,长度多达约5000个或更多核苷酸或核苷酸对或长度多达约100000个或更多核苷酸或核苷酸对。
[0589]
rna合成
[0590]
描述用于dna合成的方法可以适用于rna的合成。在一种修改中,可以修改针对本发明1和2的合成方法版本及其变体描述的合成步骤。因此,如上所述,在合成方法版本1和2及其变体的每一个中,支架多核苷酸的支持链是dna链。支架多核苷酸的合成链的引物链部分是rna链。如果存在的话,辅助链可以是rna链。如果存在,辅助链可以是dna链。
[0591]
核苷酸可以从核糖核苷-5'-o-三磷酸(ntp)合并,其可以被修饰以包含可逆终止子基团,如上所述。优选使用3'-o-修饰的核糖核苷-5'-o-三磷酸。通过rna聚合酶的作用合并修饰的核苷酸。
[0592]
因此,关于本发明1-5的合成方法版本的描述可以作必要的变通而应用于rna合成,但是如所描述的那样适用。
[0593]
图31和32描述了用于rna合成的反应方案,其分别是图6和7所示的实施例的dna合成方法版本1和2的修改。分别如图1至图5所示,本发明1和2的方法版本及其变型可以以相
同的方式进行修改。
[0594]
在任何合适的rna合成方法中,支持链、引物链、辅助链、多核苷酸连接分子和通用核苷酸的上述描述均可以作必要的变通,但可按上述方法进行修改。如前所述的裂解步骤和裂解位置可以在必要的变更后应用,因为包含通用核苷酸的支持链是dna链。在优选的实施方案中,将splintr dna连接酶用于连接步骤。
[0595]
固相合成
[0596]
根据本发明的合成方法产生的合成多核苷酸可优选使用固相或可逆固相技术合成。各种这样的技术在本领域中是已知的并且可以使用。在开始合成预定序列的新双链多核苷酸之前,可以将支架多核苷酸固定到表面上,例如平面,例如玻璃、基于凝胶的材料、或微粒例如珠子或官能化量子点的表面上。包含表面的材料本身可以与基底结合。例如,支架多核苷酸可以固定在基于凝胶的材料上,例如聚丙烯酰胺,并且其中基于凝胶的材料与支持基底如玻璃结合。
[0597]
多核苷酸可以直接或间接地固定或束缚到表面。例如,它们可以通过化学键合直接附着在表面上。它们可以通过中间表面间接地拴系在表面上,例如微粒或珠子的表面,例如在spri中或在电润湿系统中,如下所述。然后可以启动并完成合成循环,同时固定合并新合成的多核苷酸的支架多核苷酸。
[0598]
在此类方法中,可以在合并预定序列的第一核苷酸之前将双链支架多核苷酸固定到表面。因此,这种固定的双链支架多核苷酸可以充当锚,以在合成期间和之后将预定义序列的双链多核苷酸连接到表面。
[0599]
这种双链锚/支架多核苷酸的仅一条链可以在分子同一端固定在表面上。或者,双链锚/支架多核苷酸的两条链可各自在分子同一末端固定在表面上。可以提供双链锚/支架多核苷酸,其中每条链在相邻末端连接,例如通过与新合成起始位点相对的末端的发夹环,并且连接的末端可以固定在表面上(例如如图11中示意性描绘的)。
[0600]
在涉及支架多核苷酸的方法中,如本文所述,支架多核苷酸可以在以预定序列合并第一核苷酸之前附着于表面。因此,包括引物链部分和与其杂交的支持链部分的合成链可以分别附着于表面,如图11(a)和(c)所示。包括引物链部分和与其杂交的支持链部分的合成链可以在相邻末端连接,例如通过发夹环,例如在新合成起始位点的相对末端,并且连接端可以系链到表面上,如图11(b)和(d)所示。包括引物链部分和与其杂交的支持链部分的一条或另一条合成链可以分别附着于表面,如图11(e)到(h)所示。优选地,包括引物链部分和与其杂交的支持链部分的合成链附着于表面。
[0601]
平坦表面上的固相合成
[0602]
在开始合成预定义序列的新双链多核苷酸之前,合成锚/支架多核苷酸可通过本领域已知的方法合成,包含本文所述的那些,并束缚在表面上。
[0603]
可以通过常用于产生附着于平表面的核酸微阵列的方法将预形成的多核苷酸拴在表面上。例如,可以产生锚/支架多核苷酸,然后将其点样或印刷到平表面上。可以使用接触印刷技术将锚/支架多核苷酸沉积在表面上。例如,可将固体或空心尖端或针浸入包括预形成的支架多核苷酸的溶液中并与平表面接触。或者,可以将寡核苷酸吸附到微型印模上,然后通过物理接触转移到平表面。非接触印刷技术包含热印刷或压电印刷,其中包括预形成的支架多核苷酸的亚纳升尺寸微滴可以使用与喷墨和喷泡印刷中使用的方法类似的方
法从印刷尖端喷出。
[0604]
单链寡核苷酸可以直接在平表面上合成,例如使用用于产生微阵列的所谓“芯片上”方法。然后,此类单链寡核苷酸可以充当附着位点以固定预先形成的锚/支架多核苷酸。
[0605]
用于产生单链寡核苷酸的芯片上技术包括光刻法,其涉及使用通过光刻掩模引导的uv光来选择性地激活受保护的核苷酸,从而允许随后合并新的受保护的核苷酸。uv介导的脱保护和预定核苷酸偶联的循环允许原位产生具有所需序列的寡核苷酸。作为使用光刻掩模的替代方案,可以通过使用喷墨印刷技术顺序沉积核碱基并使用偶联、氧化和脱保护的循环来产生具有所需序列的寡核苷酸,从而在平表面上产生寡核苷酸(对于综述,见kosuri和church,nature methods,2014,11,499-507)。
[0606]
在本文所述的任何合成方法中,包含如下所述的涉及可逆固定的方法,表面可由任何合适的材料制成。通常,表面可包括硅、玻璃或聚合物材料。表面可包括凝胶表面,例如聚丙烯酰胺表面,例如约2%聚丙烯酰胺,任选地使用n-(5-溴乙酰基戊基)丙烯酰胺(brapa)衍生的聚丙烯酰胺表面,优选地聚丙烯酰胺表面与固体载体比如玻璃偶联。
[0607]
可逆固定
[0608]
具有预定义序列的合成多核苷酸可以根据本发明使用促进可逆固定的结合表面和结构(例如微粒和珠子)来合成。固相可逆固定(spri)方法或改良方法是本领域已知的并且可以使用(例如参见deangelis m.m.等人(1995)solid-phase reversible immobilization for the isolation of pcr products,nucleic acids research,23(22):4742-4743)。
[0609]
表面可以以微粒的形式提供,例如顺磁珠。顺磁珠可以在磁场的影响下聚集。例如,顺磁表面可以具有化学基团,例如羧基,其在适当的附着条件下将充当核酸的结合部分,如下面更详细描述的。可以在适当的洗脱条件下从这些表面洗脱核酸。微粒和珠子的表面可以提供uv敏感的聚碳酸酯。在合适的固定缓冲液存在下,核酸可以与活化表面结合。
[0610]
可使微粒和珠粒在反应溶液中自由移动,然后可逆地固定,例如通过将珠粒保持在微孔或蚀刻到表面中的凹坑中。珠子可以定位为阵列的一部分,例如通过使用附着于珠子的独特核酸“条形码”或通过使用颜色编码。
[0611]
因此,在开始合成预定义序列的新双链多核苷酸之前,可以合成根据本发明的锚/支架多核苷酸,然后可逆地固定到这样的结合表面上。通过本发明方法合成的多核苷酸可以合成,同时可逆地固定在这样的结合表面上。
[0612]
微流体技术和系统
[0613]
表面可以是电介质上电润湿系统(ewod)的一部分。ewod系统提供电介质涂覆的表面,其有助于以微滴形式的非常小的液体体积的微流体操纵(例如参见chou,w-l.等人(2015)recent advances in applications of droplet microfluidics,micromachines,6:1249-1271.)。通过电润湿技术可以可编程地在芯片上创建、移动、分区和组合液滴体积。因此,电润湿系统提供了在合成期间和之后可逆地固定多核苷酸的替代方式。
[0614]
具有预定序列的多核苷酸可以通过本文所述的方法在固相中合成,其中多核苷酸固定在ewod表面上,并且通过电润湿技术促进每个循环中所需的步骤。例如,在涉及支架多核苷酸并需要合并、裂解、连接和脱保护步骤的方法中,每个步骤所需的试剂以及用于除去用过的和不需要的试剂的任何所需洗涤步骤,可以以通过电润湿技术在电场的影响下传输
的微滴的形式提供。
[0615]
可以用于本发明的合成方法中的其它微流体平台是可用的。例如,可以使用通常用于核酸操作的基于乳液的微滴技术。在这样的系统中,微滴在通过混合两种不混溶的流体(通常是水和油)产生的乳液中形成。可以在微流体网络中可编程地创建、移动、分割和组合乳液微滴。水凝胶系统也可提供。在本文所述的任何合成方法中,微滴可以在任何合适的相容系统中操作,例如上述ewod系统和其它微流体系统,例如包括基于包括弹性体材料的组件的结构的微流体系统。
[0616]
微滴可具有任何合适的尺寸,条件是它们与本文的合成方法相容。微滴尺寸将根据所采用的特定系统和系统的相关架构而变化。因此适当地可以调整尺寸。在本文所述的任何合成方法中,液滴直径可以在约150nm到约5mm的范围内。低于1μm的液滴直径可通过本领域已知的方法验证,例如通过涉及毛细管喷射方法的技术,例如等人(nature physics,2007,3,pp737
–
742)
[0617]
中间或最终合成产物的测序。
[0618]
可以对合成或组装的中间产物或最终的多核苷酸合成产物进行测序,以进行质量控制检查,以确定所需的一种或多种多核苷酸是否已正确合成或组装。可以从固相合成平台上除去感兴趣的一种或多种多核苷酸,并通过多种已知的商业上可获得的测序技术中的任一种进行测序,例如使用牛津纳米孔技术有限公司出售的minion
tm
装置进行的纳米孔测序。测序可在固相平台本身上进行,从而无需将多核苷酸转移至单独的合成装置。测序可以方便地在相同的电润湿设备上进行,例如用于合成的ewod设备,由此合成设备包括一个或多个测量电极对。可以将包括目的多核苷酸的液滴与电极对的电极之一接触,该液滴形成液滴界面双层,其中第二液滴与电极对的第二电极接触,其中液滴双层界面在两亲性膜中包括纳米孔。例如,可以在酶的控制下使多核苷酸移位到纳米孔中,并且可以在多核苷酸通过纳米孔的过程中在电极对之间的电势差下测量通过纳米孔的离子电流。可以记录随时间变化的离子电流测量值,并用于确定多核苷酸序列。测序之前,可对多核苷酸进行一个或多个样品制备步骤,以使其最优化以进行测序,例如专利申请no.pct/gb2015/050140。可以适当使用的酶,两亲性膜和纳米孔的实例公开于专利申请no.pct/gb2013/052767和pct/gb2014/052736。可以通过样品入口将用于制备多核苷酸,纳米孔,两亲性膜等的样品所需的试剂提供给ewod设备。样品入口可以连接到试剂室。
[0619]
表面附着化学方法
[0620]
尽管寡核苷酸通常是化学连接的,但它们也可以通过间接方式例如通过亲和相互作用附着于表面。例如,寡核苷酸可以用生物素官能化并结合到用抗生物素蛋白或链酶抗生物素蛋白包被的表面上。
[0621]
为了将多核苷酸固定到表面(例如平表面)、微粒和珠子等,可以使用各种表面附着方法和化学品。表面可以被官能化或衍生化以促进附接。这种功能化是本领域中已知的。例如,表面可以用以下各者进行功能化:多组氨酸标签(六组氨酸标签、6xhis-标签、his6标签或)、ni-nta、链酶亲和素、生物素、寡核苷酸、多核苷酸(如dna、rna、pna、gna、tna或lna)、羧基、季胺基、硫醇基、叠氮基、炔基、dibo、脂质、flag-标签(flag八肽)、多核苷酸结合蛋白、肽、蛋白质、抗体或抗体片段。表面可以用与锚/支架多核苷酸特异性地结合的分子或基团进行功能化。
[0622]
适合于将多核苷酸连接到表面的化学物质的一些实例显示在图11i和图11j中。
[0623]
在本文所述的方法中的任一种中,可以通过一个或多个共价键将包括合成链的支架多核苷酸栓到共同表面,所述合成链包括引物链部分和与其杂交的支持链的部分。一个或多个共价键可以在共同表面上的官能团和支架分子上的官能团之间形成。支架分子上的官能团可以是例如胺基、硫醇基、硫代磷酸酯基或硫代酰胺基。共同表面上的官能团可以是溴乙酰基,可选地其中溴乙酰基在使用n-(5-溴乙酰基戊基)丙烯酰胺(brapa)衍生的聚丙烯酰胺表面上提供。
[0624]
在本发明的任何方法中,支架多核苷酸可以通过接头直接或间接附着于表面。可以使用任何合适的属性是生物相容且亲水的接头。
[0625]
接头可以是直链接头或支链接头。
[0626]
接头可包括烃链。烃链可包括2至约2000或更多个碳原子。烃链可包括亚烷基,例如c2至约2000或更多个亚烷基。烃链可具有通式-(ch2)
n-,其中n为2至约2000或更高。烃链可任选地被一个或多个酯基(即-c(o)-o-)或一个或多个酰胺基(即-c(o)-n(h)-)。
[0627]
可以使用选自包括以下的组的任何接头:peg、聚丙烯酰胺、聚(甲基丙烯酸2-羟乙酯)、聚-2-甲基-2-噁唑啉(pmoxa)、两性离子聚合物,例如聚(羧基甜菜碱甲基丙烯酸酯)(pcbma)、聚[n-(3-磺丙基)-n-甲基丙烯酰氧基乙基-n,n二甲基铵甜菜碱](psbma)、糖聚合物和多肽。
[0628]
接头可包含具有以下通式的聚乙二醇(peg):
[0629]-(ch 2-ch 2-o)n-,其中n为1至约600或更大。
[0630]
接头可包括具有通式为-[(ch
2-ch
2-o)
n-po
2--
o]
m-的低聚乙二醇磷酸酯单元,其中n是1到约600或更大,并且m可以是1-200或更大。
[0631]
任何上述接头可以在接头的一端连接至如本文所述的支架分子,并且在接头的另一端连接至第一官能团,其中第一官能团可以提供与表面的共价连接。第一官能团可以是例如胺基、硫醇基、硫代磷酸酯基或硫代酰胺基,如本文进一步描述的。表面可以用另外的官能团官能化以提供与第一官能团的共价键。另外的官能团可以是例如本文进一步描述的2-溴乙酰氨基。任选地,在使用n-(5-溴乙酰胺基戊基)丙烯酰胺(brapa)衍生的聚丙烯酰胺表面上提供溴乙酰基。表面上的进一步官能团可以是溴乙酰基,任选地其中溴乙酰基在使用n-(5-溴乙酰基戊基)丙烯酰胺(brapa)衍生的聚丙烯酰胺表面上提供,并且适当时第一官能团可以是例如胺基、巯基、硫代磷酸酯基或硫代酰胺基。多核苷酸附着的表面可包括凝胶。所述表面包括聚丙烯酰胺表面,例如约2%的聚丙烯酰胺,优选聚丙烯酰胺表面与固体载体如玻璃偶联。
[0632]
在本发明的任何方法中,支架多核苷酸可任选地通过合并支架多核苷酸中的支化核苷酸连接至接头。任何合适的支化核苷酸可以与任何合适的相容性接头一起使用。
[0633]
在开始本发明的合成循环之前,可以合成支架多核苷酸,其中一个或多个支化核苷酸被合并支架多核苷酸中。将一个或多个支化核苷酸合并支架多核苷酸中并因此可以连接接头的确切位置可以变化,并且可以根据需要选择。所述位置可以例如在支持链和/或合成链的末端或例如在包括发夹环的实施例中将支持链连接至合成链的环区域中。
[0634]
在支架多核苷酸的合成期间,可以将一个或多个支化核苷酸合并支架多核苷酸中,其中封闭基团阻断支化部分的反应基团。然后可以在偶联至接头的分支部分之前去除
(解封闭)封闭基团,或者如果接头包括多个单元,则将接头的第一单元(分子)去除。
[0635]
在支架多核苷酸的合成期间,可以将一个或多个支化核苷酸合并支架多核苷酸中,该支架多核苷酸具有适合用于随后的“点击化学”反应的基团,以与连接体的分支部分偶联,或者如果接头包含多个单元,与第一个单元偶联。这种基团的一个实例是乙炔基。
[0636]
一些非限制性示例性支化核苷酸如下所示。
[0637][0638]
接头可任选地包括一个或多个间隔分子(单元),例如sp9间隔基,其中第一间隔基单元被附接到分支核苷酸。
[0639]
接头可包括连接到第一间隔基团的一个或多个其它间隔基团。例如,接头可包括多个例如sp9间隔基团。将第一间隔基团连接到支化部分,然后依次加入一个或多个另外的间隔基团以延伸包括多个间隔单元的间隔基链,所述间隔单元与其间的磷酸酯基团连接。
[0640]
下面显示的是间隔基单元(sp3、sp9和sp13)的一些非限制性实例,其可以包括与分支核苷酸连接的第一间隔基单元,或者与已经与分支核苷酸连接的现有间隔基单元连接的另外的间隔基单元。
[0641][0642]
接头可包括一个或多个乙二醇单元。
[0643]
接头可包括寡核苷酸,其中多个单元是核苷酸。
[0644]
在上文描述的结构中,术语5”用于区分与支化部分连接的核苷酸的5'末端,其中5'具有本领域的普通含义。5”意指核苷酸上可以延伸接头的位置。5”的位置可能会有所不同。5”位置通常是核苷酸的核碱基中的位置。核碱基中的5”位置可以根据所需支化部分的性质而变化,如上述结构所示。
[0645]
微阵列
[0646]
本文描述的任何多核苷酸合成方法可用于制造多核苷酸微阵列(trevino,v.等人,mol.med.2007 13,pp527-541)。因此,锚或支架多核苷酸可以连接到表面上的多个可单独寻址的反应位点,并且具有预定义序列的多核苷酸可以在微阵列上原位合成。
[0647]
合成后,在每个反应区域,可以为预定序列的多核苷酸提供独特的序列。可以向锚或支架多核苷酸提供条形码序列以便于鉴定。
[0648]
除了合成预定义序列的多核苷酸的方法之外,可以使用本技术领域中常用的技术,包含本文所述的技术,进行微阵列制造。例如,可以使用已知的表面附着方法和化学方法将锚或支架多核苷酸拴在表面上,包含本文所述的那些。
[0649]
在合成预定序列的多核苷酸后,可以提供最终裂解步骤以从未束缚端移除任何不需要的多核苷酸序列。
[0650]
可以双链形式在反应位点提供预定义序列的多核苷酸。或者,在合成之后,可以分离双链多核苷酸并去除一条链,在反应位点留下单链多核苷酸。可以提供链的选择性束缚以促进所述过程。例如,在涉及支架多核苷酸的方法中,合成链可以束缚在表面上,支持链可以不被束缚,反之亦然。合成链可以具有不可裂解的接头,并且支持链可以具有可裂解的接头,反之亦然。链的分离可以通过常规方法进行,例如热处理。
[0651]
合成多核苷酸的组装
[0652]
具有通过本文所述方法合成的预定义序列并且任选地通过本文所述方法扩增的多核苷酸可以与一种或多种其它此类多核苷酸连接以产生更大的合成多核苷酸。
[0653]
可以通过本领域公知的技术实现多个多核苷酸的连接。可以裂解通过本文所描述的方法合成的第一多核苷酸和一种或多种另外的多核苷酸以产生相容的末端,并且然后通
过连接将多核苷酸连接在一起。可以通过任何合适的方法实现裂解。通常,可以在多核苷酸中产生限制性酶裂解位点,并且然后使用限制性酶执行裂解步骤,从而从任何锚/支架多核苷酸释放合成的多核苷酸。裂解位点可以被设计为锚/支架多核苷酸的一部分。或者,可以在新合成的多核苷酸内产生裂解位点,作为预定的核苷酸序列的一部分。
[0654]
多核苷酸的组装优选地使用固相方法进行。例如,在合成后,第一多核苷酸可以在远离表面固定位点的合适位置进行单一裂解。因此,第一多核苷酸将保持固定在表面上,并且单个裂解将产生与另一个多核苷酸连接相容的末端。另外的多核苷酸可以在两个合适的位置进行裂解,以在每个末端产生用于连接其他多核苷酸的相容末端,同时从表面固定中释放另外的多核苷酸。另外的多核苷酸可以与第一多核苷酸相容地连接,从而产生更大的固定化多核苷酸,其具有预定义序列并且具有与另一个额外多核苷酸连接相容的末端。因此,预选的裂解的合成多核苷酸的连接的迭代循环可以产生更长的合成多核苷酸分子。另外的多核苷酸的连接顺序将由所需的预定义序列测定。
[0655]
因此,本发明的组装方法可以允许产生长度为一个或多个mb左右的合成多核苷酸分子。
[0656]
可以使用本领域已知的装置进行本发明的组装和/或合成方法。可获得的技术和装置允许非常小体积的试剂被选择性地移动、分配并与阵列的不同位置中的其它体积组合,通常以液滴的形式,可以使用电润湿技术,例如电介质上电润湿(ewod),如上所述。例如在us8653832、us8828336、us20140197028和us20140202863中公开了可以在本发明中使用的能够操纵液滴的合适的电润湿技术和系统。
[0657]
可以通过在引物链部分和与其杂交的支持链部分中的一个或两个中提供可裂解接头来实现从固相裂解。可裂解接头可以是例如uv可裂解接头。
[0658]
涉及酶促裂解的裂解方法的实例显示在图29中。所述示意图显示了附着于表面(通过黑色金刚石结构显示)并且包括预定义序列的多核苷酸的支架多核苷酸。支架多核苷酸包括顶部和底部发夹。在每种情况下,可以使用通用核苷酸的裂解步骤裂解顶部发夹,以限定裂解位点。底部发夹可以通过限制性核酸内切酶经由设计到支架多核苷酸中的位点去除或者工程化到新合成的预定义序列的多核苷酸中。
[0659]
因此,如上所述,可以合成具有预定义序列的多核苷酸,同时固定在电润湿表面上。合成的多核苷酸可以从电润湿表面裂解下来并以液滴形式在电场的影响下移动。液滴可以在表面上的特定反应位点处组合,在所述特定反应位点其可以递送裂解的合成多核苷酸用于与其它裂解的合成多核苷酸连接。然后可以(例如通过连接)连接多核苷酸。使用这些技术,可以根据所期望的预定义序列合成并且依次连接不同多核苷酸的群体。使用这种系统,可以设计完全自动化的多核苷酸合成和组装系统。系统可以被编程为接收期望的序列、供应试剂、执行合成循环并随后根据期望的预定义序列组装期望的多核苷酸。
[0660]
系统和试剂盒
[0661]
本发明还提供了用于实施本文描述和限定的任何合成方法以及本文描述和限定的任何后续扩增和组装步骤的多核苷酸合成系统。
[0662]
通常,合成循环反应将通过将预定序列的核苷酸合并支架多核苷酸分子来进行,所述支架多核苷酸分子通过本文描述和限定的方式与表面束缚。表面可以是如本文所述和限定的任何合适的表面。
[0663]
在一个实施方案中,将预定序列的核苷酸合并支架多核苷酸分子的反应涉及在反应区域内在支架多核苷酸上进行任何合成方法。
[0664]
反应区域是支架多核苷酸分子附着的合适基底的任何区域,并且其中可以递送用于进行合成方法的试剂。
[0665]
在一个实施方案中,反应区域可以是包括单个支架多核苷酸分子的表面的单个区域,其中单个支架多核苷酸分子可以用试剂寻址。
[0666]
在另一个实施方案中,反应区域可以是包含多个支架多核苷酸分子的表面的单个区域,其中支架多核苷酸分子不能用彼此隔离的试剂单独寻址。因此,在这样的实施方案中,反应区域中的多个支架多核苷酸分子暴露于相同的试剂和条件,因此可以产生具有相同或基本相同的核苷酸序列的合成多核苷酸分子。
[0667]
在一个实施方案中,用于实施本文所述和限定的任何合成方法的合成系统可以包括多个反应区域,其中每个反应区域包括一个或多个附着的支架多核苷酸分子,并且其中每个反应区域可以与每个其它反应区域隔离地用试剂单独寻址。这种系统可以例如以阵列的形式配置,例如其中反应区域形成在基底上,通常是平面基底。
[0668]
具有包括单个反应区域或包括多个反应区域的基底的系统可包括在例如ewod系统或微流体系统内,并且系统配置成将试剂递送至反应位点。本文更详细地描述了ewod和微流体系统。例如,ewod系统可以配置成在电控制下将试剂递送到反应位点。微流体系统,例如包括微制造结构,例如由弹性体或类似材料形成的微流体系统,可以配置成在流体压力和/或抽吸控制下或通过机械方式将试剂输送到反应位点。试剂可以通过任何合适的方式递送,例如通过用作试剂递送导管的碳纳米管。可以设想任何合适的系统。
[0669]
ewod、微流体和其他系统可以配置成将任何其他所需的试剂递送至反应位点,例如用于在合成后从支架多核苷酸裂解合成的双链多核苷酸的酶,和/或用于裂解接头以从基底释放整个支架多核苷酸的试剂和/或用于在合成后扩增多核苷酸分子的试剂或其任何区域或部分,和/或用于从较小多核苷酸分子组装较大多核苷酸分子的试剂,所述较小多核苷酸分子是根据本发明的合成方法合成的。
[0670]
本发明还提供了用于实施本文所述和限定的任何合成方法的试剂盒。试剂盒可含有任何所需的试剂组合,其用于进行本文所述和限定的本发明的任何合成和/或组装方法。例如,试剂盒可包含任何一个或多个体积的反应试剂,其包含支架多核苷酸、对应于本文所述和限定的合成循环的任何一个或多个步骤的反应试剂的体积、包含含有可逆封闭基团或可逆终止子基团的核苷酸的反应试剂的体积、用于在合成之后扩增一种或多种多核苷酸分子或其任何区域或部分的反应试剂的体积、用于从根据本发明的合成方法合成的较小多核苷酸分子组装较大多核苷酸分子的反应试剂的体积、用于在合成后从支架多核苷酸裂解合成的双链多核苷酸的反应试剂的体积、以及用于裂解一个或多个接头从基底释放完整的支架多核苷酸的反应试剂的体积。
[0671]
示例性方法
[0672]
本文描述了根据本发明的示例性的非限制性合成多核苷酸或寡核苷酸分子的方法,包括所附权利要求。
[0673]
在以下根据本发明的合成多核苷酸或寡核苷酸分子的两种示例性方法及其变体中,将根据分别在图1至图5中列出的反应示意图而不是根据图6至图10中的任何图中列出
的反应示意图或实例部分中的描述来解释对合成方法版本1和2的引用。在下面的实施例部分中,在图6至10中任何一个上列出的反应示意图及其描述为基于与本发明的方法相比被修改的反应方案提供了对本发明方法的说明性支持。
[0674]
在下文所描述的每一示例性方法中,在每一步骤中所描述的结构可在适当时借助于附图标记来参考特定图。因此,下文的文本中的附图标记与图1到5中的附图标记对应。然而,此类参考并不希望限于图式中展示的结构,并且相关结构的描述对应于如本文中所提供的全部描述,包含但不限于特定说明的那些。
[0675]
下面描述本发明的两种非限制性示例性方法,在本文中称为本发明1和2的合成方法版本(分别参见例如图1和2)。在这些示例性方法的每种方法的步骤(1)中,提供了一个支架多核苷酸(参见图1和2的步骤1中所示的结构)(101、201),其包括与互补支持链(参见图1和2中的每个图的步骤1中描绘的结构中被标记为“a”的链)杂交的合成链(参见图1和2中的每个图的步骤1中描绘的结构中被标记为“b”的链)。
[0676]
支架多核苷酸是双链的并且提供支持结构以修改合成多核苷酸的区域,因为它是从头合成的。支架多核苷酸包括合成链,所述合成链包括引物链部分(参见图1和图2中的每个图的步骤1中描绘的结构中被标记为“b”的链的点线部分)和辅助链部分(参见图1和图2中的每个图的步骤1中描绘的结构中被标记为“b”的链的虚线部分)。合成链的引物链部分和辅助链部分都与互补的支持链杂交。
[0677]
包含引物链部分的支架多核苷酸的末端包含平端,即在任一链中均没有突出的核苷酸。
[0678]
在方法的步骤(2)中,进行连接步骤(102、202),其中将多核苷酸连接分子连接至双链支架多核苷酸。多核苷酸连接分子包含预定核苷酸序列的第一核苷酸。多核苷酸连接分子包含支持链和与支持链杂交的辅助链。多核苷酸连接分子包含平端互补连接端,即在任一链中均没有突出的核苷酸。平端互补连接端与双链支架多核苷酸的平端互补。多核苷酸连接分子的支持链在互补的连接端包含预定核苷酸序列的第一核苷酸。预定义核苷酸序列的第一核苷酸是多核苷酸连接分子在互补连接端的支持链的末端核苷酸。预定核苷酸序列的第一核苷酸是可连接的核苷酸,并与支架多核苷酸的支持链的末端核苷酸连接。连接后,通过在双链支架多核苷酸的平端连接至双链支架多核苷酸的支持链,将预定核苷酸序列的第一核苷酸合并双链支架多核苷酸中。
[0679]
在这两种方法版本及其变体的每一个中,多核苷酸连接分子的支持链在互补连接端还包含一个通用核苷酸(在图1和图2所示的结构中标记为“un”),这将有助于在裂解步骤中进行裂解。通用核苷酸的作用将从以下每种方法的详细描述中显而易见。
[0680]
提供多核苷酸连接分子的辅助链在互补连接端的末端核苷酸,使得辅助链不能被连接至合成链的引物链部分,即,其被提供为不可连接的核苷酸。这通常通过提供没有磷酸基的辅助链的末端核苷酸来实现,即它以核苷形式提供。或者,可以使用5'-保护的核苷,在5'位置具有不可连接基团的核苷,例如5'-脱氧核苷或5'-氨基核苷,或任何其他合适的不可连接的核苷酸或核苷。
[0681]
因此,在将多核苷酸连接分子的支持链与双链支架多核苷酸的支持链连接时,在合成链的引物链部分和辅助链之间在合成链中提供了单链断裂或“缺口”。
[0682]
将多核苷酸连接分子连接至双链支架多核苷酸后,形成双链支架多核苷酸,其包
含新合并的第一核苷酸,用于促进裂解步骤中的裂解的通用核苷酸和“缺口”。
[0683]
由于该循环的预定序列的第一核苷酸与双链支架多核苷酸的支持链的末端核苷酸连接,因此必须在连接步骤之前为双链支架多核苷酸的支持链的末端核苷酸提供附接的磷酸基团或其他可连接基团,以使双链支架多核苷酸的支持链的末端核苷酸充当连接酶的底物。
[0684]
如本文中更详细描述的,在与本文所述的版本1和2及其变体有关的某些示例性方法中,可以在将预定序列的第二核苷酸合并到该合成循环中的步骤之前移除辅助链,例如通过变性和从先前与其杂交的支持链中释放。
[0685]
在合成的第一循环的上下文中,术语“预定序列的第一核苷酸”不必理解为意指预定序列的第一核苷酸。本文所述的方法涉及具有预定序列的双链多核苷酸的合成,并且可以在开始第一个合成循环之前在支架多核苷酸中预合成提供预定序列的一部分。在本文中,预定义序列的术语“一个”第一核苷酸可以意指预定义序列的“任何”核苷酸。
[0686]
合成链的引物链部分的末端提供了引物位点,该引物位点是用于附接预定序列的第二核苷酸的位点,该第二核苷酸通过具有延伸寡核苷酸或具有单个核苷酸的多核苷酸分子的能力的酶被附接/合并到合成链中。这样的酶通常是核苷酸转移酶或聚合酶。任何合适的可以使用本文进一步定义和/或技术人员已知的酶。因此,该酶将起作用以延伸引物链部分的末端核苷酸。因此,这一末端核苷酸将通常定义引物链部分的3'端,例如允许通过聚合酶或转移酶延伸,所述聚合酶或转移酶在5'到3'方向上催化延伸。包括引物链部分的合成链的相对末端将因此通常定义合成链的5'端,并且与合成链的5'端相邻的支持链的末端核苷酸将因此通常定义支持链的3'端。
[0687]
位于单链断裂的位点处的合成链的辅助链部分的末端核苷酸通常将限定辅助链部分的5'末端,并因此合成链的辅助链部分的相对末端通常将限定合成链的3'末端。
[0688]
在合并第二核苷酸的步骤(步骤3;103、203)中,第二核苷酸设置有可逆终止子基团团(在图1和2中的每个图的步骤3中被描绘为合并的核苷酸的小三角形),其防止通过酶进一步延伸。因此,在步骤(3)中仅合并单核苷酸。
[0689]
可以使用包括任何合适的可逆终止子基团的核苷酸。具有可逆终止子基团的优选核苷酸是3
′-
o-烯丙基-dntp和/或3
′-
o-叠氮甲基-dntp或本文进一步描述的其他基团。
[0690]
从本文中所定义的各种方法的描述中将显而易见的是,术语“预定序列的第二核苷酸”不应理解为是指从包括预定序列的一条链中的线性序列中的第一核苷酸开始的下一核苷酸,而仅是在整个合成的双链多核苷酸的上下文中的预定序列的“一”另外的核苷酸。在本文定义的本发明的特定和非限制性方法版本1和2及其某些变体的情况下,一个循环中的每个“第一核苷酸”将在相同的顺序中顺序连接至上一个循环的“第一核苷酸”核酸链,从而每个循环将第一链顺序延伸一核苷酸。在每个循环中,“第二核苷酸”将与“第一核苷酸”配对,并且一个循环中的每个“第二核苷酸”将在同一核酸链中与前一个循环的“第二核苷酸”相邻地顺序引入,从而延伸第二个链按每个循环顺序一核苷酸排列。因此,当合成循环完成时,合成的双链多核苷酸分子将包含由每个循环的连接的第一核苷酸限定的一条链的预定序列,以及由每个循环的结合的第二核苷酸限定的相反链的预定序列。两条链的序列都必须预先定义,并由使用者在每个合成循环中选择的第一和第二核苷酸的身份决定。在本文所述的方法版本1和2中,假定每个循环的第一和第二核苷酸形成一核苷酸对,如果使
用者选择每个循环的第二核苷酸与每个循环的第一核苷酸天然互补,则最终合成链将完全互补。如果使用者选择某些循环的第二核苷酸与那些循环的各个第一核苷酸不互补,那么最终的合成链将不是完全互补的。然而,在任何一种情况下,最终的合成链均包含序列,该序列在合成的双链多核苷酸整体上是预定的。
[0691]
在合并第二核苷酸的步骤(步骤3)之后,然后裂解支架多核苷酸(步骤4、104、204)。裂解引起多核苷酸连接分子从支架多核苷酸中释放,并且该循环的第一核苷酸保留在裂解的支架多核苷酸的支持链上,并与该循环的第二核苷酸配对,该第二核苷酸与该支架的合成链连接裂解的支架多核苷酸。裂解会引起辅助链(如果存在)释放,并在裂解前立即与支持链杂交,并释放包含通用核苷酸的支持链。裂解因此将裂解的双链支架多核苷酸留在原位,该双链支架多核苷酸在裂解位点包含裂解的支持链的裂解末端和包含裂解前切口位点的合成链的引物链部分的末端,其中裂解的双链支架多核苷酸包含该循环的第一核苷酸作为支持链的裂解末端的末端核苷酸,与该循环的第二核苷酸配对作为合成链的引物链部分的末端核苷酸。
[0692]
在该方法的步骤(5)中,进行脱保护步骤以从预定核苷酸序列(105、205)的合并的第二核苷酸中去除可逆终止基。脱保护步骤可以可选地在裂解步骤(步骤4)之前进行,在这种情况下,脱保护步骤定义为步骤(4)(图1和2的步骤4;104、204),并且裂解步骤定义为步骤(5)(图1和2的步骤5;105、205)。
[0693]
进行包括上述相同步骤的合成的迭代循环,以产生合成的多核苷酸。
[0694]
具体方法在下文更详细地描述。
[0695]
合成方法版本1
[0696]
参考图1,在本发明的合成方法的第一特定的非限制性示例性形式中,提供了双链支架多核苷酸(图1的步骤1;101)。该双链支架多核苷酸包含支持链和与其杂交的合成链。合成链包含引物链部分。所述双链支架多核苷酸具有至少一个平端,其中所述至少一个平端包括引物链部分的末端和与其杂交的支持链的末端。支持链的末端核苷酸能够充当连接酶的底物,并且优选包含磷酸基团或其他可连接基团。
[0697]
在该方法的步骤(2)中,将多核苷酸连接分子(参见图1上部的右上方所示的结构)连接至支架多核苷酸。连接将预定序列的第一核苷酸合并到支架多核苷酸的支持链中(图1的步骤2;102)。
[0698]
在该方法的步骤(3)中,在具有利用单核苷酸延伸寡核苷酸或多核苷酸分子的能力的酶的作用下,将预定核苷酸序列的第二核苷酸添加到合成链的引物链部分的末端。这样的酶通常是核苷酸转移酶或聚合酶(图1的步骤3;103)。第二核苷酸具有可逆终止子基团,其防止通过酶进一步延伸。因此,在步骤(3)中仅合并单核苷酸。
[0699]
然后在每个合成循环中,在由支持链中包含通用核苷酸的序列限定的裂解位点裂解支架多核苷酸(步骤4;104)。在方法版本1中,裂解包括在通用核苷酸之后立即在靠近引物链部分/远离辅助链的方向上裂解支持链,即,在由通用核苷酸占据的位置和支持链中沿靠近引物链部分/远离辅助链的方向的下一核苷酸位置之间裂解支持链。支架多核苷酸的裂解(步骤4)引起多核苷酸连接分子从支架多核苷酸的释放和该循环的第一核苷酸的保留保留在裂解的支架多核苷酸的第一链上,并与该循环的第二核苷酸配对,寡核苷酸连接至合成链的引物链部分。支架多核苷酸的裂解(步骤4)引起支架多核苷酸的辅助链丢失(如果
存在并在裂解前立即与支持链杂交),以及通用核苷酸从支架多核苷酸的丧失。裂解将裂解的双链支架多核苷酸留在原位,该双链支架多核苷酸在裂解位点包含裂解的支持链的裂解末端和包含裂解前切口位点的合成链的引物链部分的末端。裂解引起在裂解位点的平端裂解的双链支架多核苷酸,在任一链中都没有突出,并且第一和第二核苷酸为核苷酸对。
[0700]
在该方法的步骤(5)中,进行脱保护步骤以从新合并的核苷酸中移除终止子基团。脱保护步骤可以可替代地在裂解步骤之前进行,在这种情况下,脱保护步骤定义为步骤(4),而裂解步骤定义为步骤(5),如图1所示(分别为104和105)。
[0701]
步骤1-提供支架多核苷酸
[0702]
在本发明的合成方法的示例性版本1中,在步骤(1)(101)中提供了双链支架多核苷酸。提供了双链支架多核苷酸,其包含合成链和与其杂交的支持链,其中所述合成链包含引物链部分。支持链的末端核苷酸与引物链部分的末端核苷酸配对,从而形成平端。支持链的末端核苷酸能够充当连接酶的底物,并且包含可连接基团,优选磷酸基团。
[0703]
步骤2-将多核苷酸连接分子连接至支架多核苷酸并合并预定序列的第一核苷酸
[0704]
在该方法的步骤(2)中,在平端连接反应中,通过连接酶的作用将双链多核苷酸连接分子连接(102)至支架多核苷酸。
[0705]
多核苷酸连接分子包含与之杂交的支持链和辅助链。多核苷酸连接分子进一步包含互补的连接端,其在支持链中包含通用核苷酸和预定序列的第一核苷酸。
[0706]
多核苷酸连接分子的互补的连接端被构造成使得支持链的末端核苷酸是在任何给定的合成循环中被合并到支架多核苷酸中的预定序列的第一核苷酸。支持链的末端核苷酸与辅助链的末端核苷酸配对。支持链的末端核苷酸,即该循环的预定序列的第一核苷酸,在支持链中占据核苷酸位置n。位置n是指在合并第二核苷酸后与该循环的预定序列的第二核苷酸相对的核苷酸位置,第一和第二核苷酸由此形成核苷酸对。在图1中,预定序列的第一核苷酸被描述为腺苷,步骤(3)中预定序列的第二核苷酸被描述为胸腺嘧啶,辅助链的末端核苷酸被描述为胸腺嘧啶。腺苷和胸腺嘧啶仅用于说明。辅助链的第一和第二核苷酸以及末端核苷酸可以是使用者选择的任何合适的核苷酸。描绘为x的核苷酸也可以是使用者选择的任何合适的核苷酸。
[0707]
通常,在互补连接端的支持链的末端核苷酸将定义多核苷酸连接分子的互补连接端的支持链的3'末端。
[0708]
配置多核苷酸连接分子的辅助链的末端核苷酸,使其不能与多核苷酸链中的另一个多核苷酸连接(纯标记为“t”的位置仅用于说明图上部右上方的结构)1)。该核苷酸称为不可连接的末端核苷酸。通常,该末端核苷酸将缺少磷酸基团,即它将是核苷。通常,如上文所描述,此辅助链的末端核苷酸将定义辅助链的5'端。
[0709]
在多核苷酸连接分子的互补连接端中,支持链中的通用核苷酸被定位成使得它是支持链的次末端核苷酸,与辅助链的次末端核苷酸配对,并且在支持链中占据核苷酸位置n+1。位置n+1是指在支持链中相对于位置n在互补连接端远端的方向上的下一核苷酸位置。
[0710]
互补的连接端被配置为使得当经受合适的连接条件时,它将与支架多核苷酸的平末端相容地连接。在多核苷酸连接分子的支持链与支架多核苷酸连接后,第一核苷酸被合并到支架多核苷酸中。由于多核苷酸连接分子的辅助链的末端核苷酸是不可连接的核苷酸,因此将阻止连接酶连接合成链的辅助链和引物链部分,从而在辅助链与合成链的引物
链部分之间产生单链断裂或“缺口”。
[0711]
多核苷酸连接分子与支架多核苷酸的连接延长了步骤(1)的双链支架多核苷酸的支持链的长度,并且其中预定核苷酸序列的第一核苷酸被合并到支架多核苷酸的支持链中。
[0712]
支持链的连接可以通过任何合适的方式进行。连接通常可以并且优选地通过具有连接酶活性的酶来进行。例如,可以用t3dna连接酶或t4dna连接酶或其功能性变体或其等同物或本文所述的其他酶进行连接。这样的酶的使用将引起在合成链中维持单链断裂,因为提供了辅助链的末端核苷酸,使得其不能充当连接酶的底物,例如由于不存在末端磷酸基团或存在不可连接的封闭基团。
[0713]
在将多核苷酸连接分子连接至支架多核苷酸之后,将预定核苷酸序列的第一核苷酸称为占据核苷酸位置n,将通用核苷酸称为占据核苷酸位置n+1,并且在连接之前为靠近引物链部分的支架多核苷酸的支持链的末端核苷酸被称为占据核苷酸位置n-1。核苷酸位置n-1是指在支持链中相对于位置n在引物链部分近端/辅助链远端的方向上的下一核苷酸位置。
[0714]
步骤3-合并预定序列的第二核苷酸
[0715]
在该方法的步骤(3)中,在将多核苷酸连接分子连接到支架多核苷酸之后,然后通过延伸引物链部分将预定序列的第二核苷酸合并到合成链中。
[0716]
引物链部分的延伸可通过任何合适的酶的作用来实现,该酶具有延伸具有单核苷酸的寡核苷酸或多核苷酸分子的能力。这样的酶通常是核苷酸转移酶或聚合酶。可以使用本文进一步定义的或本领域技术人员已知的任何合适的酶。
[0717]
如果在紧接引物链部分延伸之前,在支架多核苷酸中存在辅助链,尤其是如果使用聚合酶,则该酶可起到“侵入”辅助链并置换辅助链末端核苷酸的作用。然后,并入的第二核苷酸将占据先前被辅助链的置换末端核苷酸占据的位置(参见图1下部中间所示的结构)。在某些实施方案中,可以在延伸/合并步骤(3)之前除去辅助链,在这种情况下,酶可以接近并延伸合成链的引物链部分的末端核苷酸,而无需置换辅助链或其一部分。
[0718]
在步骤(3)中并入的预定序列的第二核苷酸包括阻止通过酶进一步延伸的可逆终止子基团,或包括阻止通过酶进一步延伸的任何其他类似功能。可以使用本文进一步定义的或本领域技术人员已知的任何合适的可逆终止子基团或功能。
[0719]
将其并入支架多核苷酸的引物链部分中后,该循环的预定序列的第二核苷酸与该循环的预定序列的第一核苷酸配对以形成该循环的核苷酸对。核苷酸对可以是本文进一步定义的任何合适的核苷酸对。
[0720]
步骤4——裂解
[0721]
在该方法的步骤(4)中,支架多核苷酸在裂解位点被裂解(104)。裂解位点由包含支持链中的通用核苷酸的序列限定。裂解引起支架多核苷酸中的双链断裂。支架多核苷酸的裂解(步骤4)引起辅助链的丢失(如果存在并且在裂解前立即与支持链杂交)以及包含通用核苷酸的支持链的丢失。
[0722]
支架多核苷酸的裂解从而从支架多核苷酸释放多核苷酸连接分子,但是引起该循环的第一核苷酸保留在裂解的支架多核苷酸的第一链上并与该循环的第二核苷酸配对。支架多核苷酸的裂解将裂解的钝端双链支架多核苷酸留在原位,该多核苷酸平端双链支架多
核苷酸在支持链的末端包含预定序列的第一核苷酸,并且在合成链的引物链部分的末端包含预定序列的第二核苷酸,其中,第一和第二核苷酸形成核苷酸对。
[0723]
在该示例性方法中,合成链已经具有单链断裂或“缺口”,因此仅需要裂解支持链即可在支架多核苷酸中提供双链断裂。此外,如前所述,在该示例性方法版本中,裂解产生了平端裂解的双链支架多核苷酸,在合成链或支持链中均没有突出端,并且通用核苷酸在支持链之前位于支持链中的位置n+1裂解步骤。为了在通用核苷酸在支持链中位于n+1位置时获得这种平末端裂解的双链支架多核苷酸,将支持链在相对于通用核苷酸的特定位置裂解。当支架多核苷酸的支持链在核苷酸位置n+1和n之间被裂解时,多核苷酸连接分子从支架多核苷酸中释放(参见图1上部左上方所示的结构),除了该循环保留在与裂解的支架多核苷酸的支持链相连的支架多核苷酸中。
[0724]
磷酸基团应继续在裂解位点连接至裂解的支架多核苷酸的支持链的末端核苷酸。这确保了在下一合成循环的连接步骤中,可以将裂解的支架多核苷酸的支持链连接至多核苷酸连接分子的支持链。进行裂解使得裂解的支架多核苷酸的支持链的末端核苷酸保留可连接基团,优选末端磷酸基团,并且因此不需要进行磷酸化步骤。
[0725]
因此,在方法版本1中,通用核苷酸在步骤(2),(3)和(4)在支持链中位于位置n+1,并且在步骤(4)将支持链在核苷酸位置n+1和n之间裂解。
[0726]
优选地,通过在核苷酸位置n+1和n之间的磷酸二酯键的裂解来裂解支持链(支持链的第一个磷酸二酯键相对于通用核苷酸的位置,沿远离辅助链/靠近引物链的方向)。
[0727]
可以通过裂解核苷酸位置n+1和n之间的磷酸二酯键的一个酯键来裂解支持链。
[0728]
优选地,通过相对于核苷酸位置n+1裂解第一酯键来裂解支持链。这将具有在裂解位置处在裂解的支架多核苷酸的支持链上保留末端磷酸酯基团的作用。
[0729]
当通用核苷酸占据位置n+1时,可以采用任何合适的机制来实现在核苷酸位置n+1和n之间的支持链的裂解。
[0730]
如上所述,在核苷酸位置n+1和n之间裂解支持链可以通过酶的作用进行。
[0731]
如上所述,核苷酸位置n+1和n之间的支持链的裂解可以作为两步过程进行。
[0732]
第一裂解步骤可包括从支持链除去通用核苷酸,从而在位置n+1处形成无碱基位点,第二裂解步骤可包括在无基位点在位置n+1和n之间裂解支持链。
[0733]
实施例2中描述了一种以上述方式在包含通用核苷酸的序列所定义的裂解位点裂解支持链的机制。实施例2中描述的裂解机理是示例性的,并且可以采用其他机理,只要实现上述平端裂解的双链支架多核苷酸即可。
[0734]
在两步裂解过程的第一个裂解步骤中,将通用核苷酸从支持链上移出,同时保留糖-磷酸主链的完整性。这可以通过酶的作用来实现,所述酶可以从双链多核苷酸中特异性地切除单个通用核苷酸。在示例性裂解方法中,通用核苷酸是肌苷,并且通过酶的作用从支持链切除肌苷,从而形成无碱基位点。在示例性裂解方法中,酶是3-甲基腺嘌呤dna糖基化酶,特别是人类烷基腺嘌呤dna糖基化酶(haag)。可以使用其它酶、分子或化学品,条件是形成无碱基位点。核苷酸切除酶可以是催化尿嘧啶从多核苷酸释放的酶,例如尿嘧啶-dna糖基化酶(udg)。
[0735]
在两步裂解过程的第二个裂解步骤中,通过单链断裂,在无碱基位点裂解支持链。在示例性方法中,支持链通过作为碱如naoh的化学物质的作用裂解。或者,可以使用有机化
学品,如n,n'-二甲基乙二胺。或者,可以使用具有无碱基位点裂解酶活性的酶,例如ap核酸内切酶1、核酸内切酶iii(nth)或核酸内切酶viii。可以使用其他酶、分子或化学品,条件是支持链在如上所述的无碱基位点处被裂解。
[0736]
因此,在其中通用核苷酸在步骤(1)和(2)处在支持链的位置n+1处并且支持链在位置n+1和n之间裂解的实施方案中,可以用核苷酸切除酶进行第一裂解步骤。这种酶的实例是3-甲基腺嘌呤dna糖基化酶,例如人类烷基腺嘌呤dna糖基化酶(haag)。第二裂解步骤可以用作为碱的化学物质如naoh进行。第二步可以用具有无碱基位点裂解活性的有机化学品如n,n'-二甲基乙二胺进行。第二步骤可以用具有脱碱基位点裂解酶活性的酶如核酸内切酶viii或核酸内切酶iii进行。
[0737]
如上所述,在核苷酸位置n+1和n之间的支持链的裂解也可以作为一步裂解过程来进行。可用于任何此类方法的酶的实例包括核酸内切酶iii、核酸内切酶viii。可以在任何这样的方法中使用的其他酶包括裂解8-氧代鸟苷的酶,例如甲酰胺基嘧啶dna糖基化酶(fpg)和8-氧代鸟嘌呤dna糖基化酶(hogg1)。
[0738]
步骤5——脱保护
[0739]
在本发明的该示例性方法形式以及所有形式中,为了允许将下一个核苷酸并入下一个合成循环,必须将可逆终止子基团从第一个核苷酸中除去(脱保护步骤;105)。这可在第一循环的各个阶段执行。在裂解步骤(4)之后,它可以作为方法的步骤(5)进行。然而,脱保护步骤可以在步骤(3)中引入第二核苷酸之后并且在裂解步骤(4)之前进行,在这种情况下,脱保护步骤被定义为步骤(4),裂解步骤被定义为步骤(5),如图1所示(分别为104和105)。无论执行哪个保护基步骤,都应在步骤(3)之后首先去除酶和残留的未结合的第二核苷酸,以防止在同一合成循环中多次合并第二核苷酸。
[0740]
可以通过本领域技术人员已知的任何合适的方式从第一核苷酸中移除可逆终止子基团。举例来说,可通过使用化学物质(例如,三(羧乙基)膦(tcep))来进行移除。
[0741]
进一步循环
[0742]
在完成第一合成循环之后,可以使用相同的方法步骤进行第二合成循环和其他合成循环。
[0743]
(在步骤6中)提供前一循环的步骤(4)和(5)的裂解产物作为用于下一合成循环的双链支架多核苷酸。
[0744]
在下一个合成循环和每个其他合成循环的步骤(6)中,将另一个双链多核苷酸连接分子连接至上一个循环的步骤(4)和(5)的裂解产物。所述多核苷酸连接分子可以以与上述对于先前循环的步骤(2)相同的方式构造,不同之处在于,所述多核苷酸连接分子包含进一步的合成循环的第一核苷酸。多核苷酸连接分子可以以与上述步骤(2)相同的方式连接至前一循环的步骤(4)和(5)的裂解产物。
[0745]
在下一个和每个进一步合成循环的步骤(7)中,通过引入进一步合成循环的第二核苷酸,进一步延长了双链支架多核苷酸合成链的引物链部分的末端。核苷酸转移酶,聚合酶或其他酶的作用。可以按照与上述步骤(3)相同的方式合并进一步合成循环的第二核苷酸。
[0746]
在合成的下一循环和每个进一步循环的步骤(8)中,在裂解位点裂解连接的支架多核苷酸。裂解引起支架多核苷酸中的双链断裂。支架多核苷酸的裂解(步骤8)引起辅助链
的丢失(如果存在并且在裂解前立即与支持链杂交)以及包含通用核苷酸的支持链的丢失。支架多核苷酸的裂解从而从支架多核苷酸释放另外的多核苷酸连接分子,但是引起该另外的循环的第一核苷酸的保留保留在裂解的支架多核苷酸的支持链上,并与该另外的循环的第二核苷酸配对。支架多核苷酸的裂解将裂解的钝端双链支架多核苷酸留在原位,该支架在支架多核苷酸的支持链的末端包含另一个循环的第一核苷酸,在末端包含另一个循环的第二核苷酸支架多核苷酸的合成链的引物链部分的“末端”。步骤(8)的裂解可以以与上述步骤(4)相同的方式进行。
[0747]
在步骤(9)中,将可逆终止基团从另一个循环的第二核苷酸中除去(脱保护步骤;109)。如上面对于第一循环所述,这可以在各个阶段执行。在裂解步骤(8)之后,它可以作为方法的步骤(9)进行。或者,可以在合并步骤(7)之后且在裂解步骤(8)之前的任何步骤进行脱保护步骤,在这种情况下,将脱保护步骤定义为步骤(8),并将裂解步骤定义为步骤(9),如图1所示(分别为109和110)。通过在下一个循环和随后的循环中去除可逆终止基团来进行脱保护可以如上文关于第一合成循环所述进行。
[0748]
如上文所描述根据需要重复合成循环多次,以合成具有预定核苷酸序列的双链多核苷酸。
[0749]
合成方法版本2
[0750]
参考图2,在本发明的合成方法的第二特定的非限制性示例性形式中,提供了双链支架多核苷酸(图2的步骤1;201)。该双链支架多核苷酸包含支持链和与其杂交的合成链。合成链包含引物链部分。所述双链支架多核苷酸具有至少一个平端,其中所述至少一个平端包括引物链部分的末端和与其杂交的支持链的末端。支持链的末端核苷酸能够充当连接酶的底物,并且优选包含磷酸基团或其他可连接基团。
[0751]
在该方法的步骤(2)中,将多核苷酸连接分子(参见图2上部的右上方所示的结构)连接至支架多核苷酸。连接将预定序列的第一核苷酸合并到支架多核苷酸的支持链中(图2的步骤2;202)。
[0752]
在该方法的步骤(3)中,在具有利用单核苷酸延伸寡核苷酸或多核苷酸分子的能力的酶的作用下,将预定核苷酸序列的第二核苷酸添加到合成链的引物链部分的末端。这种酶通常是核苷酸转移酶或聚合酶(图2的步骤3;203)。第二核苷酸具有可逆终止子基团,其防止通过酶进一步延伸。因此,在步骤(3)中仅合并单核苷酸。
[0753]
然后在每个合成循环中,在由支持链中包含通用核苷酸的序列限定的裂解位点裂解支架多核苷酸(步骤4;204)。在方法版本2中,裂解包括在紧接引物链部分/远离辅助链的方向上在紧接通用核苷酸的核苷酸位置的核苷酸之后立即裂解支持链。支架多核苷酸的裂解(步骤4)引起多核苷酸连接分子从支架多核苷酸的释放和该循环的第一核苷酸的保留保留在裂解的支架多核苷酸的第一链上,并与该循环的第二核苷酸配对,寡核苷酸连接至合成链的引物链部分。支架多核苷酸的裂解(步骤4)引起支架多核苷酸的辅助链丢失(如果存在并在裂解前立即与支持链杂交),以及通用核苷酸从支架多核苷酸的丧失。裂解将裂解的双链支架多核苷酸留在原位,该双链支架多核苷酸在裂解位点包含裂解的支持链的裂解末端和包含裂解前切口位点的合成链的引物链部分的末端。裂解产生在裂解位点的平端裂解的双链支架多核苷酸,在任一链中均没有突出端,并且第一和第二核苷酸为核苷酸对。
[0754]
在该方法的步骤(5)中,进行脱保护步骤以从新合并的核苷酸中移除终止子基团。
脱保护步骤也可以在裂解步骤之前进行,在这种情况下,脱保护步骤定义为步骤(4),裂解步骤定义为步骤(5),如图2所示(分别为204和205)。
[0755]
步骤1-提供支架多核苷酸
[0756]
在本发明的合成方法的示例性版本2中,在步骤(1)中提供了双链支架多核苷酸(201)。提供了双链支架多核苷酸,其包含合成链和与其杂交的支持链,其中所述合成链包含引物链部分。支持链的末端核苷酸与支持链的末端核苷酸配对,从而形成平端。支持链的末端核苷酸能够充当连接酶的底物,并且包含可连接基团,优选磷酸基团。
[0757]
步骤2-将多核苷酸连接分子连接至支架多核苷酸并合并预定序列的第一核苷酸
[0758]
在该方法的步骤(2)中,在平端连接反应中,通过连接酶的作用将双链多核苷酸连接分子连接(202)至支架多核苷酸。
[0759]
多核苷酸连接分子包含与之杂交的支持链和辅助链。多核苷酸连接分子进一步包含互补的连接端,其在支持链中包含通用核苷酸和预定序列的第一核苷酸。
[0760]
多核苷酸连接分子的互补的连接端被构造成使得支持链的末端核苷酸是在任何给定的合成循环中被合并到支架多核苷酸中的预定序列的第一核苷酸。支持链的末端核苷酸与辅助链的末端核苷酸配对。支持链的末端核苷酸,即该循环的预定序列的第一核苷酸,在支持链中占据核苷酸位置n。位置n是指在合并第二核苷酸后与该循环的预定序列的第二核苷酸相对的核苷酸位置,第一和第二核苷酸由此形成核苷酸对。在图2中,预定序列的第一核苷酸被描述为腺苷,步骤(3)中预定序列的第二核苷酸被描述为胸腺嘧啶,辅助链的末端核苷酸被描述为胸腺嘧啶。腺苷和胸腺嘧啶仅用于说明。辅助链的第一和第二核苷酸以及末端核苷酸可以是任何合适的核苷酸。描绘为x的核苷酸也可以是使用者选择的任何合适的核苷酸。
[0761]
通常,在互补连接端的支持链的末端核苷酸将定义多核苷酸连接分子的互补连接端的支持链的3'末端。
[0762]
配置多核苷酸连接分子的辅助链的末端核苷酸,使其不能与多核苷酸链中的另一个多核苷酸连接(纯标记为“t”的位置仅用于说明图上部右上方的结构)2)。该核苷酸称为不可连接的末端核苷酸。通常,该末端核苷酸将缺少磷酸基团,即它将是核苷。通常,如上文所描述,此辅助链的末端核苷酸将定义辅助链的5'端。
[0763]
在多核苷酸连接分子的互补连接端中,支持链中的通用核苷酸被定位成使得其在支持链中占据核苷酸位置n+2,并与辅助链中的配偶体核苷酸配对。位置n+2是指在支持链中相对于互补连接端远端的方向上的位置n的第二核苷酸位置。
[0764]
互补的连接端被配置为使得当经受合适的连接条件时,它将与支架多核苷酸的平末端相容地连接。在多核苷酸连接分子的支持链与支架多核苷酸连接后,第一核苷酸被合并到支架多核苷酸中。由于多核苷酸连接分子的辅助链的末端核苷酸是不可连接的核苷酸,因此将阻止连接酶连接合成链的辅助链和引物链部分,从而在辅助链与合成链的引物链部分之间产生单链断裂或“缺口”。
[0765]
多核苷酸连接分子与支架多核苷酸的连接延长了步骤(1)的双链支架多核苷酸的支持链的长度,并且其中预定核苷酸序列的第一核苷酸被合并到支架多核苷酸的支持链中。
[0766]
支持链的连接可以通过任何合适的方式进行。连接通常可以并且优选地通过具有
连接酶活性的酶来进行。例如,可以用t3 dna连接酶或t4 dna连接酶或其功能变体或等同物或本文进一步描述的其他酶进行连接。这样的酶的使用将引起在合成链中维持单链断裂,因为提供了辅助链的末端核苷酸,使得其不能充当连接酶的底物,例如由于不存在末端磷酸基团或存在不可连接的封闭基团。
[0767]
在将多核苷酸连接分子连接至支架多核苷酸之后,将预定核苷酸序列的第一核苷酸称为占据核苷酸位置n,将通用核苷酸称为占据核苷酸位置n+2,并且在连接之前为靠近引物链部分的支架多核苷酸的支持链的末端核苷酸被称为占据核苷酸位置n-1。核苷酸位置n-1是指在支持链中相对于位置n在引物链部分近端/辅助链远端的方向上的下一核苷酸位置。
[0768]
步骤3-合并预定序列的第二核苷酸
[0769]
在该方法的步骤(3)中,在将多核苷酸连接分子连接到支架多核苷酸之后,然后通过延伸引物链部分将预定序列的第二核苷酸合并到合成链中。
[0770]
引物链部分的延伸可通过任何合适的酶的作用来实现,该酶具有延伸具有单核苷酸的寡核苷酸或多核苷酸分子的能力。这样的酶通常是核苷酸转移酶或聚合酶。可以使用本文进一步定义的或本领域技术人员已知的任何合适的酶。
[0771]
如果在紧接引物链部分延伸之前,在支架多核苷酸中存在辅助链,尤其是如果使用聚合酶,则该酶可起到“侵入”辅助链并置换辅助链末端核苷酸的作用。然后,合并的第二核苷酸将占据先前被辅助链的置换末端核苷酸占据的位置(参见图2下部中间所示的结构)。在某些实施方案中,可以在延伸/合并步骤(3)之前除去辅助链,在这种情况下,酶可以接近并延伸合成链的引物链部分的末端核苷酸,而无需置换辅助链或其一部分。
[0772]
在步骤(3)中并入的预定序列的第二核苷酸包括阻止通过酶进一步延伸的可逆终止子基团,或包括阻止通过酶进一步延伸的任何其他类似功能。可以使用本文进一步定义的或本领域技术人员已知的任何合适的可逆终止子基团或功能。
[0773]
将其并入支架多核苷酸的引物链部分中后,该循环的预定序列的第二核苷酸与该循环的预定序列的第一核苷酸配对以形成该循环的核苷酸对。核苷酸对可以是本文进一步定义的任何合适的核苷酸对。
[0774]
步骤4-裂解
[0775]
在该方法的步骤(4)中,连接的支架多核苷酸在裂解位点被裂解(204)。裂解位点由包含支持链中的通用核苷酸的序列限定。裂解引起支架多核苷酸中的双链断裂。支架多核苷酸的裂解(步骤4)引起辅助链的丢失(如果存在并且在裂解前立即与支持链杂交)以及包含通用核苷酸的支持链的丢失。
[0776]
支架多核苷酸的裂解从而从支架多核苷酸释放多核苷酸连接分子,但是引起该循环的第一核苷酸保留在裂解的支架多核苷酸的第一链上并与该循环的第二核苷酸配对。支架多核苷酸的裂解将裂解的钝端双链支架多核苷酸留在原位,该多核苷酸平端双链支架多核苷酸在支持链的末端包含预定序列的第一核苷酸,并且在合成链的引物链部分的末端包含预定序列的第二核苷酸,其中,第一和第二核苷酸形成核苷酸对。
[0777]
在该示例性方法中,合成链已经具有单链断裂或“缺口”,因此仅需要裂解支持链即可在支架多核苷酸中提供双链断裂。此外,如之前在该示例性方法版本中所述,裂解产生了平端裂解的双链支架多核苷酸,其在合成链或支持链中均未突出,并且通用核苷酸在支
持链之前位于支持链中的n+2位裂解步骤。为了在通用核苷酸位于支持链中的n+2位置时获得这种平末端裂解的双链支架多核苷酸,在相对于通用核苷酸的特定位置裂解支持链。当支架多核苷酸的支持链在核苷酸位置n+1和n之间被裂解时,多核苷酸连接分子从支架多核苷酸中释放(参见图1上部左上方所示的结构),除了该循环保留在与裂解的支架多核苷酸的支持链相连的支架多核苷酸中。
[0778]
磷酸基团应继续在裂解位点连接至裂解的支架多核苷酸的支持链的末端核苷酸。这确保了在下一合成循环的连接步骤中,可以将裂解的支架多核苷酸的支持链连接至多核苷酸连接分子的支持链。进行裂解使得裂解的支架多核苷酸的支持链的末端核苷酸保留可连接基团,优选末端磷酸基团,并且因此不需要进行磷酸化步骤。
[0779]
因此,在方法版本1中,通用核苷酸在步骤(2)、(3)和(4)在支持链中占据位置n+2,并且在步骤(4)将支持链在核苷酸位置n+1和n之间裂解。
[0780]
优选地,通过在核苷酸位置n+1和n之间的磷酸二酯键的裂解来裂解支持链(支持链的第一个磷酸二酯键相对于通用核苷酸的位置,沿远离辅助链/靠近引物链的方向)。
[0781]
可以通过裂解核苷酸位置n+1和n之间的磷酸二酯键的一个酯键来裂解支持链。
[0782]
优选地,通过相对于核苷酸位置n+1裂解第一酯键来裂解支持链。这将具有在裂解位置处在裂解的支架多核苷酸的支持链上保留末端磷酸酯基团的作用。
[0783]
当通用核苷酸占据位置n+2时,可以采用任何合适的机制来实现在核苷酸位置n+1和n之间的支持链的裂解。
[0784]
如上所述,在核苷酸位置n+1和n之间裂解支持链可以通过酶的作用进行。
[0785]
如上所述,当通用核苷酸占据支持链中的位置n+2时,在核苷酸位置n+1和n之间裂解支持链可以通过酶例如核酸内切酶v的作用来进行。
[0786]
在实例3中以类似的方式描述了在由包含在支持链中占据n+2位的通用核苷酸的序列限定的裂解位点处在核苷酸位置n+1和n之间裂解支持链的一种机制。所描述的机制为示例性的并且可采用其它机制,其条件是实现上文所描述的裂解布置。
[0787]
在此示例性机制中,采用核酸内切酶。在示例性方法中,酶是核酸内切酶v。可以使用其他酶,分子或化学物质,只要当通用核苷酸在支持链中的位置n+2处将支持链在核苷酸位置n+1和n之间裂解时即可。
[0788]
步骤5-脱保护
[0789]
在本发明的该示例性方法形式以及所有形式中,为了允许将下一个核苷酸并入下一个合成循环,必须将可逆终止子基团从第一个核苷酸中除去(脱保护步骤;205)。这可在第一循环的各个阶段执行。在裂解步骤(4)之后,它可以作为方法的步骤(5)进行。然而,脱保护步骤可以在步骤(3)中引入第二核苷酸之后并且在裂解步骤(4)之前进行,在这种情况下,脱保护步骤被定义为步骤(4),并且裂解步骤被定义为步骤(5),如图2所示(分别为204和205)。无论执行哪个保护基步骤,都应在步骤(3)之后首先去除酶和残留的未结合的第二核苷酸,以防止在同一合成循环中多次合并第二核苷酸。
[0790]
可以通过本领域技术人员已知的任何合适的方式从第一核苷酸中移除可逆终止子基团。举例来说,可通过使用化学物质(例如,三(羧乙基)膦(tcep))来进行移除。
[0791]
进一步循环
[0792]
在完成第一合成循环之后,可以使用相同的方法步骤进行第二合成循环和其他合
成循环。
[0793]
(在步骤6中)提供前一循环的步骤(4)和(5)的裂解产物作为用于下一合成循环的双链支架多核苷酸。
[0794]
在下一个合成循环和每个其他合成循环的步骤(6)中,将另一个双链多核苷酸连接分子连接至上一个循环的步骤(4)和(5)的裂解产物。所述多核苷酸连接分子可以以与上述对于先前循环的步骤(2)相同的方式构造,不同之处在于,所述多核苷酸连接分子包含进一步的合成循环的第一核苷酸。多核苷酸连接分子可以以与上述步骤(2)相同的方式连接至前一循环的步骤(4)和(5)的裂解产物。
[0795]
在下一个和每个进一步合成循环的步骤(7)中,通过引入进一步合成循环的第二核苷酸,进一步延长了双链支架多核苷酸合成链的引物链部分的末端。核苷酸转移酶,聚合酶或其他酶的作用。可以按照与上述步骤(3)相同的方式合并进一步合成循环的第二核苷酸。
[0796]
在合成的下一循环和每个进一步循环的步骤(8)中,在裂解位点裂解连接的支架多核苷酸。裂解引起支架多核苷酸中的双链断裂。支架多核苷酸的裂解(步骤8)引起辅助链的丢失(如果存在并且在裂解前立即与支持链杂交)以及包含通用核苷酸的支持链的丢失。支架多核苷酸的裂解从而从支架多核苷酸释放另外的多核苷酸连接分子,但是引起该另外的循环的第一核苷酸的保留保留在裂解的支架多核苷酸的支持链上,并与该另外的循环的第二核苷酸配对。支架多核苷酸的裂解将裂解的钝端双链支架多核苷酸留在原位,该支架在支架多核苷酸的支持链的末端包含另一个循环的第一核苷酸,在末端包含另一个循环的第二核苷酸支架多核苷酸的合成链的引物链部分的“末端”。步骤(8)的裂解可以以与上述步骤(4)相同的方式进行。
[0797]
在步骤(9)中,将可逆终止基团从另一个循环的第二核苷酸中除去(脱保护步骤;209)。如上面对于第一循环所述,这可以在各个阶段执行。在裂解步骤(8)之后,它可以作为方法的步骤(9)进行。或者,可以在合并步骤(7)之后且在裂解步骤(8)之前的任何步骤进行脱保护步骤,在这种情况下,将脱保护步骤定义为步骤(8),并将裂解步骤定义为步骤(9),如图2所示(分别为209和210)。通过在下一个循环和随后的循环中去除可逆终止基团来进行脱保护可以如上文关于第一合成循环所述进行。
[0798]
如上文所描述根据需要重复合成循环多次,以合成具有预定核苷酸序列的双链多核苷酸。
[0799]
合成方法版本2的变体
[0800]
除了上述方法之外,还提供了合成方法版本2的变体,其中,除了以下变化之外,该方法以与上述合成方法版本2相同的方式执行。
[0801]
在第一个循环的连接步骤(步骤2)中,多核苷酸连接分子的互补连接端的结构应使通用核苷酸位于支持链上的核苷酸位置n+2+x处,并与配偶体核苷酸配对。在互补连接端从辅助链的末端核苷酸除去2+x位的辅助链;其中核苷酸位置n+2是支持链中相对于互补连接端远端方向上核苷酸位置n的第二核苷酸位置。
[0802]
在第一循环的裂解步骤(步骤4)中,通用核苷酸代替地在支架多核苷酸的支持链中占据核苷酸位置n+2+x,其中,核苷酸位置n+2是支持链中沿靠近辅助链/远离引物链部分的方向相对于核苷酸位置n的第二核苷酸位置;并且在位置n+1和n之间裂解支架多核苷酸
的辅助链。
[0803]
在第二个循环的连接步骤(步骤6)和所有其他循环的连接步骤中,多核苷酸连接分子的互补连接端的结构应使通用核苷酸在支持链中占据核苷酸位置n+2+x,并且与在互补连接端从辅助链的末端核苷酸去除2+x位的配偶体核苷酸配对;其中核苷酸位置n+2是支持链中相对于互补连接端远端方向上核苷酸位置n的第二核苷酸位置。
[0804]
最后,在第二个循环的裂解步骤(步骤8)和所有其他循环的裂解步骤中,通用核苷酸代替地在支架多核苷酸的支持链中占据核苷酸位置n+2+x,其中核苷酸位置n+2为支持链中第二核苷酸位置相对于核苷酸链位置n在辅助链近侧/引物链部分的远侧;支架多核苷酸的支持链在位置n+1和n之间裂解。
[0805]
在所有这些变型方法中,x是1至10或更大的整数,并且其中x是在步骤(2)、(4)、(6)和(8)中相同的整数。
[0806]
与版本2的合成方法一样,在基于版本2的变体方法中,应注意的是,在任何给定的循环中,连接后以及合并和裂解步骤中,该循环的第一核苷酸在支持链中占据的核苷酸位置定义为核苷酸位置n,并且在完成给定的合成循环后,被裂解的支架多核苷酸的支持链中该循环的第一核苷酸占据的位置被定义为下一合成循环中的核苷酸位置n-1。
[0807]
因此,在合成方法版本2的这些特定变体中,裂解位点相对于核苷酸位置n的位置保持恒定,并且通用核苷酸相对于核苷酸位置n的位置通过在靠近辅助链/远离引物链部分的方向上移动通用核苷酸的位置而增加由为x选择的数目确定的核苷酸位置的数目。
[0808]
这些变型方法的示意图在图3中提供,其中脱保护步骤示为步骤(4),裂解步骤示为步骤(5)。如上所述,可以切换执行这些步骤的顺序。
[0809]
合成方法版本1和2的变体,每个循环包含两个以上的核苷酸
[0810]
本发明进一步提供了另外的另外的变体方法,其中每个循环合并两个以上的核苷酸,包括基于上述特定版本1和2及其变体的其他另外的变体方法。除了可以进行以下修改之外,可以如上所述执行这些其他变型方法中的任何一种。
[0811]
在步骤(2)中,多核苷酸连接分子具有互补的连接端,该互补的连接端包含第一循环的预定序列的第一核苷酸,并且还包含第一循环的预定序列的一个或多个其他核苷酸。然后将多核苷酸连接分子连接至支架多核苷酸。第一循环的预定序列的第一核苷酸是互补连接端的支持链的末端核苷酸。互补连接端优选地被构造成使得第一循环的预定序列的第一和其他核苷酸包含线性核苷酸序列,其中,序列中的每个核苷酸占据支持链中沿远离互补连接端的方向的下一核苷酸位置。
[0812]
在步骤(3)中,通过核苷酸转移酶或聚合酶的作用,通过合并第一循环的预定序列的第二核苷酸来延长双链支架多核苷酸的合成链的引物链部分的末端。酶,其中引物链部分的末端通过核苷酸转移酶或聚合酶的作用,通过并入第一循环的预定序列的一个或多个其他核苷酸而进一步延伸,其中,第一循环的第二和另外的核苷酸中的每一个包括可逆终止子基团,所述可逆终止子基团阻止通过酶进一步延伸,并且其中,在每次进一步延伸之后,在合并下一核苷酸之前在脱保护步骤(步骤4)中从核苷酸中移除可逆终止子基团。
[0813]
多核苷酸连接分子的互补连接端的结构使得在裂解之前的步骤(4)中,通用核苷酸在支持链中占据一个位置,该位置在互补连接端远端的方向上位于第一和其他核苷酸的核苷酸位置之后。
[0814]
在裂解后的步骤(4)中,将第一循环的预定序列的第一、第二和其他核苷酸保留在裂解的支架多核苷酸中。
[0815]
在步骤(6)中,多核苷酸连接分子具有互补的连接端,该互补的连接端包含另一循环的预定序列的第一核苷酸,并且还包含另一循环的预定序列的一个或多个其他核苷酸。如步骤(2)中所述,然后将多核苷酸连接分子连接至支架多核苷酸。如在步骤(2)中一样,另一个循环的预定序列的第一核苷酸是互补连接端的支持链的末端核苷酸。互补的连接端优选地被结构化,使得另外的循环的预定序列的第一和另外的核苷酸包含线性核苷酸序列,其中该序列中的每核苷酸在互补连接端远端的方向上占据支持链中的下一核苷酸位置。
[0816]
在步骤(6)中,通过核苷酸转移酶或聚合酶的作用,通过并入进一步循环的预定序列的第二核苷酸来延长双链支架多核苷酸的合成链的引物链部分的末端。酶,其中引物链部分的末端通过核苷酸转移酶或聚合酶的作用通过并入进一步循环的预定序列的一个或多个另外的核苷酸而进一步延伸,其中第二个和第二个所述另一个循环的其他核苷酸包含可逆终止子基团,所述可逆终止子基团阻止通过酶进一步延伸,并且其中,在每次进一步延伸后,在合并下一核苷酸之前,将可逆终止基团从核苷酸上除去。
[0817]
在裂解后的步骤(8)中,将另外的循环的预定序列的第一、第二和另外的核苷酸保留在裂解的支架多核苷酸中。
[0818]
在这些变体方法中,裂解总是在最后的脱保护步骤(步骤4和8)之后按照步骤(5)和(9)进行,除了在任何给定循环中合并的最后的另外核苷酸的可逆终止基团可以选择地是在裂解位点裂解连接的支架多核苷酸的步骤后,将其从最后的另一核苷酸中除去。
[0819]
在这些变体方法中,在任何给定的合成循环中,在裂解之前,在支持链中,预定序列的第一核苷酸占据的位置可以称为位置n,预定序列的第一另外核苷酸在支持链中占据的位置可以被称为n+1位,并且通用核苷酸占据的位置可以被称为n+2+x,其中x是从零到10或更大的整数,其中x是如果支持链仅包含一个另外的核苷酸,则为零;如果支持链仅包含另外的两核苷酸,则x为一个,依此类推。
[0820]
在图4中提供了这些变体方法的图解表示,其中脱保护步骤示为步骤(4),裂解步骤示为步骤(5)。如上所述,可以相对于待合并的最后一核苷酸来改变可以执行这些步骤的顺序。
[0821]
在这些变体方法中的任何一种中,多核苷酸连接分子的互补连接端的结构使得在裂解之前的步骤(4)和(8)中,通用核苷酸在支持链中占据一个位置,该位置是支持链中沿远离互补连接端的方向在第一个和其他核苷酸的核苷酸位置之后的下一个核苷酸位置,并且在最后一个核苷酸占据的位置与通用核苷酸占据的位置之间裂解支持链。
[0822]
在这些变体方法中的任何一种中,多核苷酸连接分子的互补连接端被交替地构造,使得在裂解之前的步骤(4)和(8)中,通用核苷酸在支持链中占据一个位置,该位置是支持链中沿远离互补连接端的方向在第一个和其他核苷酸的核苷酸位置之后的再下一个核苷酸位置,并且可替代地,在由最后一个另外的核苷酸占据的位置与由支持链中的下一核苷酸占据的位置之间裂解支持链。
[0823]
应当理解,就通用核苷酸相对于核苷酸位置n的位置而言,为了在合成的第一和其他循环中修改其他核苷酸的合并,除了每个第一和第二核苷酸之外,可能需要对多核苷酸连接分子的互补连接端的构型进行常规修改。在本文描述和定义的本发明的所有方法中,
核苷酸位置n始终是支持链中的核苷酸位置,该核苷酸位置在合并之前或之后与任何给定循环的预定序列的第二核苷酸相反或将与之相反。因此,本领域技术人员能够容易地对通用核苷酸的位置和相对于核苷酸位置n的裂解位点的选择进行常规修饰,以修改任何方法版本和其变体,以适应对合成的第一和另外的循环的另外的核苷酸的合并。
[0824]
实例
[0825]
以下实例对用于合成根据本发明的多核苷酸或寡核苷酸的方法以及用于所述方法的示例性构建体提供支持。实例不限制本发明。
[0826]
除实施例13以外,以下实施例描述了根据反应方案的合成方法,该反应方案与本发明的合成方法有关但不在本发明的合成方法的范围内。
[0827]
实施例证明了执行合成反应的能力,所述合成反应包括以下步骤:向支架多核苷酸的合成链中添加预定序列的核苷酸,在由通用核苷酸限定的裂解位点裂解支架多核苷酸并连接多核苷酸连接分子,其包含用于预定序列的添加核苷酸的配偶体核苷酸,以及用于创建裂解位点以用于下一合成循环的新通用核苷酸。本发明的方法以修改的方式结合了这些步骤。因此,除了实施例13以外,以下实施例提供了对本文定义的本发明方法的说明性支持。实施例13提供了与根据本发明方法,例如本发明1和2的合成方法版本及其变体的合并步骤,通过终止子x dna聚合酶合并3'-o-修饰的-dntps的数据(图1至5)。
[0828]
在以下实例中,以及在对应的图12至图50中,对例如合成方法“版本1、2和3”或“版本1、2或3”的引用根据图6、图7和图8并非根据图1至图5中任何一个所阐述的反应示意图或本文的描述来解释。将根据本发明的合成方法解释实施例13和图51。特别地,根据本发明1和2的合成方法及其变体和相关的反应示意图,其显示在图1-5中,更具体地讲,包括这些方法的步骤3。
[0829]
实例1:不存在辅助链的合成。
[0830]
该实施例描述了使用4个步骤合成多核苷酸:合并在部分双链dna上进行3'-o-修饰的dntps的裂解,连接和脱保护,第一步是在通用核苷酸(在这种情况下是肌苷)的对面进行。
[0831]
步骤1:合并
[0832]
第一步描述了通过dna聚合酶的酶促合并将3
’-
o-保护的单核苷酸受控的添加到寡核苷酸中(图12a)。
[0833]
材料和方法
[0834]
材料
[0835]
1.3
’-
o-修饰的dntp根据博士论文中描述的方案内部合成:jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis,columbia university,2008。用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[0836]
2.寡核苷酸是内部设计的并从sigma-aldrich获得(图12h)。以100μm的浓度制备储备溶液。
[0837]
3.使用终止子ix dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。然而,可以使用任何可以合并修饰的dntp的dna聚合酶。
[0838]
测试了两种类型的可逆终止剂:
[0839][0840]
方法
[0841]
1.将2μl的10x缓冲液(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4、0.1%x-100,ph 8.8,新英格兰生物实验室(new england biolabs))与12.25μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[0842]
2.将0.5μl的10μm引物(合成链)(5pmol,1当量)(seq id no:1,图12h)和0.75μl的10μm模板(支持链)(6pmol,1.5当量)(seq id no:2,图12h)加入到反应混合物中。
[0843]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[0844]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰生物实验室)。然而,可以使用任何可以合并修饰的dntp的dna聚合酶。
[0845]
5.将反应物在65℃培育20分钟。
[0846]
6.将反应物通过加入tbe-尿素样品缓冲液(novex)停止。
[0847]
7.将反应物在聚丙烯酰胺凝胶(15%)上用tbe缓冲液分离,并通过chemidoc mp成像系统(biorad)进行可视化。
[0848]
凝胶电泳和dna可视化:
[0849]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[0850]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[0851]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[0852]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[0853]
结果
[0854]
来自new england biolabs的定制工程化therminator ix dna聚合酶是一种高效的dna聚合酶,能够整合与通用核苷酸相对的3
′-
o-修饰的dntp,例如肌苷(图12b-c)。
[0855]
相对肌苷有效地合并发生在65℃的温度(图12d-e)。
[0856]
与肌苷相对的3
′-
o-修饰的dttp的结合需要存在mn
2+
离子(图12f-g)。成功转换在图12c、e、g和h中以粗体标记。
[0857]
结论
[0858]
3-o-改性dttp与肌苷相对地合并可用new england biolabs的定制工程化终止子ix dna聚合酶在mn
2+
离子存在下和在65℃的温度下以特别高的效率实现。
[0859]
步骤2:裂解
[0860]
第二步描述了用haag/endo viii或haag/化学碱两步裂解多核苷酸(图13a)。
[0861]
材料和方法
[0862]
材料
[0863]
1.实例1中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图13(e)的序列表)。
[0864]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[0865]
方法
[0866]
使用以下程序对寡核苷酸进行裂解反应:
[0867]
1.用移液管(gilson)将41μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[0868]
2.然后将5μl的10x反应缓冲液neb(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4、0.1%x-100,ph 8.8)加入同一eppendorf管中。
[0869]
3.将1靗每种寡核苷酸(图13e);模板(seq id no:3)或任何荧光标记的长寡链,具有t(seq id no:4)的引物和对照(seq id no:5),均为5pmol添加到同一试管中。
[0870]
4.将1μl人类烷基腺嘌呤dna糖基化酶(haag)neb(10单位/μl)添加到同一试管中。
[0871]
5.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[0872]
6.通常在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[0873]
在环境条件下纯化。使用下面概述的方案纯化样品混合物:
[0874]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[0875]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[0876]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph 7.5和80%乙醇),并以6000rpm离心1分钟。
[0877]
4.将流过液丢弃并将旋转柱以13000rpm离心另外的1分钟以去除残余的pe缓冲液。
[0878]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[0879]
6.对于dna洗脱,将50μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)加入柱膜中心并在室温下静置1分钟。
[0880]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[0881]
使用以下方案测定纯化的dna浓度:
[0882]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[0883]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[0884]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[0885]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[0886]
使用以下程序进行产生的无碱基位点的裂解:
[0887]
1.将2μl(10-100ng/μl)dna加入无菌的1.5ml eppendorf管中。
[0888]
2.在同一试管中加入40μl(0.2m)naoh或1.5μl endo viii neb(10单位/μl)和5μl的10x反应缓冲液neb(10mm tris-hcl,75mm nacl,1mm edta,ph 8@25℃)并通过再悬浮温和混合和在13000rpm离心5秒。
[0889]
3.将得到的混合物在室温下培育5分钟以使naoh处理样品,同时将endo viii反应混合物在37℃下培育1小时。
[0890]
4.培育时间过去之后,将反应混合物用如上面概述的纯化方案的步骤1-7纯化。
[0891]
凝胶电泳和dna可视化:
[0892]
1.将5μl的dna和tbe-尿素样品缓冲液(novex)加入无菌1.5ml eppendorf管中,并使用加热热块(eppendorf)加热至95℃,持续2分钟。
[0893]
2.然后dna混合物加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[0894]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[0895]
4.采用使用cy3 leds的chemidoc mp(biorad)进行检测和dna在凝胶中的可视化。可视化和分析在image lab 2.0平台上进行。
[0896]
结果和结论
[0897]
没有辅助链的裂解反应显示裂解的dna与未裂解的dna的低百分率产率为约7%:93%(图13b-d)。
[0898]
裂解结果显示,在该具体实施例中,并且基于所用的特定试剂,与正对照相比,在不存在辅助链的情况下获得了低产率的裂解dna。此外,与endoviii裂解相比,使用化学碱来裂解无碱基位点的时间消耗较少。
[0899]
步骤3:连接
[0900]
第三步描述了在不存在辅助链的情况下用dna连接酶连接多核苷酸。图14中显示了示意图。
[0901]
材料和方法
[0902]
材料
[0903]
1.实例1中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图14c的序列表)。
[0904]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释至100μm的储备浓度。
[0905]
方法
[0906]
使用以下程序进行寡核苷酸的连接反应:
[0907]
1.用移液管(gilson)将16μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[0908]
2.然后将10μl的2x快速连接反应缓冲液neb(132mm tris-hcl、20mm mgcl2、2mm二硫苏糖醇、2mm atp、15%聚乙二醇(peg6000)和ph 7.6,在25℃)加入到相同的eppendorf管中。
[0909]
3.加入1μl每种寡核苷酸(图14c);tamra或任何荧光标记的磷酸酯链(seq id no:
7),具有t(seq id no:8)和肌苷链(seq id no:9)的引物到同一试管中,均为5pmol。
[0910]
4.将1μl的quick t4 dna连接酶neb(400单位/μl)加入同一试管中。
[0911]
5.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[0912]
6.通常在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[0913]
7.使用如上所述的纯化步骤1-7中概述的方案纯化反应混合物。
[0914]
使用以下方案测定纯化的dna浓度:
[0915]
1.加入2μl灭菌蒸馏水(elgaveolia)到基座平衡nanodrop one(thermo scientific)。
[0916]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[0917]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one,然后在掩蔽后重复步骤2。
[0918]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[0919]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照上述步骤5-8中的程序可视化。没有引入条件或试剂的变化。
[0920]
结果和结论
[0921]
在该具体实施例中,并且基于所用的特定试剂,在没有辅助链的情况下,在室温(24℃)下用dna连接酶(在该特定情况下为快速t4 dna连接酶)连接寡核苷酸引起减少量的连接产物(图14b)。
[0922]
实例2利用辅助链的版本1化学方法.
[0923]
该实施例描述了使用4个步骤合成多核苷酸:合并
[0924]
从切口位点裂解3
’-
o-修饰的dntp,连接和脱保护,其中第一步与通用核苷酸相对地进行,在该特定情况下通用核苷酸为肌苷。该方法使用辅助链,其提高了连接和裂解步骤的效率。
[0925]
步骤1:合并
[0926]
第一步描述了通过dna聚合酶的酶促合并将3
’-
o-保护的单核苷酸受控的添加到寡核苷酸中(图15a)。
[0927]
材料和方法
[0928]
材料
[0929]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis。columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[0930]
2.寡核苷酸是内部设计的并从sigma-aldrich获得。以100μm的浓度制备储备溶液。寡核苷酸显示在图15b中。
[0931]
3.使用终止子ix dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。
[0932]
测试了两种类型的可逆终止剂:
[0933][0934]
方法
[0935]
1.2μl 10x缓冲液(20mm的tris-hcl,10mm的(nh4)2so4,10mm kcl,2mm mgso4、0.1%x-100,ph 8.8,新英格兰实验室)与10.25μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[0936]
2.将0.5μl的10μm引物(5pmol,1当量)(seq id no:10,图15(b)中的表)、0.75μl的10μm模板(6pmol,1.5当量)(seq id no:11,图15(b)中的表)、2μl的10μm辅助链(seq id no:12,图15(b)中的表)加入到反应混合物中。
[0937]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[0938]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰生物实验室)。
[0939]
5.将反应物在65℃培育20分钟。
[0940]
6.将反应物通过加入tbe-尿素样品缓冲液(novex)停止。
[0941]
7.将反应物在聚丙烯酰胺凝胶(15%)上用tbe缓冲液分离,并通过chemidoc mp成像系统(biorad)进行可视化。
[0942]
凝胶电泳和dna可视化:
[0943]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[0944]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[0945]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[0946]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[0947]
可以根据上述方案研究合并步骤。
[0948]
步骤2:裂解
[0949]
第二步描述了用haag/endo viii或haag/化学碱(x2)两步裂解多核苷酸(图16a)。
[0950]
材料和方法
[0951]
材料
[0952]
1.实例2中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图16f序列)。
[0953]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[0954]
方法
[0955]
使用以下程序进行寡核苷酸的裂解反应:
[0956]
1.用移液管(gilson)将41μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[0957]
2.然后将5μl的10x反应缓冲液neb(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4,0.1%x-100,ph 8.8)加入同一eppendorf管中。
[0958]
3.将1μl每种寡核苷酸(图16f);模板(seq id no:13)或任何荧光标记的长寡链,具有t(seq id no:14)的引物,对照(seq id no:15)和辅助链(seq id no:16),均为5pmol添加到同一试管中,。
[0959]
4.将1μl人类烷基腺嘌呤dna糖基化酶(haag)neb(10单位/μl)加入同一试管中。
[0960]
5.在使用替代碱的反应中,加入1μl的人类烷基腺嘌呤dna糖基化酶(haag)neb(100单位/μl)。
[0961]
6.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[0962]
7.通常在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[0963]
在环境条件下纯化。使用下面概述的方案纯化样品混合物:
[0964]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[0965]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[0966]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph7.5和80%乙醇),并以6000rpm离心1分钟。
[0967]
4.将流过液丢弃并将旋转柱以13000rpm离心另外的1分钟以去除残余的pe缓冲液。
[0968]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[0969]
6.对于dna洗脱,将50μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)加入柱膜中心并在室温下静置1分钟。
[0970]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[0971]
使用以下方案测定纯化的dna浓度:
[0972]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[0973]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[0974]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[0975]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[0976]
使用以下程序进行产生的无碱基位点的裂解:
[0977]
1.将2μl(10-100ng/μl)dna加入无菌的1.5ml eppendorf管中。
[0978]
2.在同一试管中加入40μl(0.2m)naoh或1.5μl endo viii neb(10单位/μl)和5μl的10x反应缓冲液neb(10mm tris-hcl,75mm nacl,1mm edta,ph 8@25℃)并通过再悬浮温和混合和在13000rpm离心5秒。
[0979]
3.将得到的混合物在室温下培育5分钟以使0.2m naoh处理样品,同时将endo viii反应混合物在37℃下培育1小时。
[0980]
4.培育时间过去之后,将反应混合物用如上面所述的纯化方案的步骤1-7纯化。
[0981]
使用以下程序进行使用替代碱性化学品的产生的无碱基位点的裂解:
[0982]
1.将1μl(10-100ng/μl)dna加入无菌的1.5ml eppendorf管中。然后将在室温下用乙酸溶液sigma(99.8%)缓冲至ph7.4的2μl的n,n'-二甲基乙二胺sigma(100mm)加入同一试管中。
[0983]
2.将20μl无菌蒸馏水(elga veolia)加入管中,通过重悬浮轻轻混合和在13000rpm离心5秒。
[0984]
3.将所得混合物在37℃下培育20分钟。
[0985]
4.培育时间过去之后,将反应混合物用如上面所述的纯化方案的步骤1-7纯化。
[0986]
凝胶电泳和dna可视化:
[0987]
1.将5μl的dna和tbe-尿素样品缓冲液(novex)加入无菌1.5ml eppendorf管中,并使用加热热块(eppendorf)加热至95℃,持续2分钟。
[0988]
2.然后dna混合物加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[0989]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[0990]
4.采用使用cy3 leds的chemidoc mp(biorad)进行检测和dna在凝胶中的可视化。可视化和分析在image lab 2.0平台上进行。
[0991]
结果
[0992]
通过haag dna糖基化酶在包含通用核苷酸(在该特定情况下为肌苷)的裂解位点处的裂解效率从辅助链不存在时的10%显著增加至辅助链存在下的50%(图16b)。haag和核酸内切酶viii以比haag和naoh(50%)更低的效率(10%)裂解肌苷。在使用带切口的dna的所述系统中,使用0.2m naoh的化学裂解显示对于ap位点的裂解优于核酸内切酶viii(图16c)。在中性ph下的温和n,n'-二甲基乙二胺具有如0.2m naoh高的效率裂解无碱基位点,并因此与核酸内切酶viii和naoh相比是优选的(图16d-e)。
[0993]
结论
[0994]
对于裂解含有肌苷的dna,评估了三种方法。在实例2中对于dna裂解研究了一种完全的酶促方法-haag/核酸内切酶viii,以及两种结合化学和酶促裂解的方法-haag/naoh和haag/二甲基乙胺。
[0995]
haag/naoh结果显示,与不存在辅助链(10%)相比,在辅助链存在下裂解dna的产率(50%)高得多。在这些具体的实施例中,并且基于所用的特定试剂,辅助链增加了dna裂解的产率。
[0996]
在辅助链存在下,与naoh(50%)相比,使用核酸内切酶viii作为naoh的替代物的酶促裂解效率较低(10%)。
[0997]
包含替代的温和化学碱n,n'-二甲基乙二胺引起ap位点的高裂解效率,与naoh一样有效,并且与添加10x haag酶一起对裂解的dna具有显著增加(参见图16e)。
[0998]
步骤3:连接
[0999]
第三步描述了在辅助链存在下用dna连接酶连接多核苷酸。图17a中示出了图解说明。
[1000]
材料和方法
[1001]
材料
[1002]
1.寡核苷酸是内部设计的并由sigma aldrich合成(参见图17d序列)。
[1003]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1004]
方法
[1005]
使用以下程序进行寡核苷酸的连接反应:
[1006]
1.用移液管(gilson)将16μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[1007]
2.然后将10μl的2x快速连接反应缓冲液neb(132mm tris-hcl、20mm mgcl2、2mm二硫苏糖醇、2mm atp、15%聚乙二醇(peg6000)和ph 7.6,在25℃)加入到相同的eppendorf管中。
[1008]
3.将1μl每种寡核苷酸(图17d);tamra或任何荧光标记的磷酸酯链(seq id no:18),具有t(seq id no:19)和肌苷链(seq id no:20)和辅助链(seq id no:21)的引物,均为5pmol加入到同一试管中。
[1009]
4.将1μl的quick t4 dna连接酶neb(400单位/μl)加入同一试管中。
[1010]
5.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[1011]
6.通常在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[1012]
7.使用如上所述的纯化步骤1-7中概述的方案纯化反应混合物。
[1013]
使用以下方案测定纯化的dna浓度:
[1014]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1015]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1016]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1017]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[1018]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照上述步骤5-8中的程序可视化。没有引入条件或试剂的变化。
[1019]
结果和结论
[1020]
在该具体实施例中,并且基于所使用的特定试剂,在没有辅助链的情况下观察到降低的连接活性(图17b),而在辅助链存在下连接以高效率进行(图17c)并且产物以高产率形成。
[1021]
实例3用辅助链的版本2化学法.
[1022]
该实施例描述了使用4个步骤合成多核苷酸:在部分双链dna上合并3
’-
o-修饰的dntp;在第一步合并与天然互补核苷酸相对地进行的情况下裂解、连接和脱保护,该天然互补核苷酸位于支持链中与通用核苷酸相邻,在该特定情况下通用核苷酸为肌苷。
[1023]
步骤1:合并
[1024]
材料和方法
[1025]
材料
[1026]
第一步描述了通过dna聚合酶的酶促合并将3
’-
o-保护的单核苷酸受控的添加到寡核苷酸中(图18a)。
[1027]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis。columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1028]
2.寡核苷酸是内部设计的并从sigma-aldrich获得(图18j)。以100μm的浓度制备储备溶液。
[1029]
3.使用终止子ix dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。
[1030]
所有dntp的3'-o-叠氮甲基可逆终止子独立测试其合并:
[1031][1032]
方法
[1033]
1.将2μl的10x缓冲液(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4、0.1%x-100,ph 8.8,新英格兰实验室)与12.25μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1034]
2.将0.5μl的10μm引物(5pmol,1当量)(seq id no:22,图18j)和0.75μl的10μm模板-a/g/t/c(6pmol,1.5当量)(seq id nos:23-26,图18j)以及1μl的10μm辅助链-t/c/a/g(10pmol,2当量)(seq id nos:27-30,图18j)加入反应混合物中。
[1035]
3.加入3
’-
o-修饰的-dttp/dctp/datp/dgtp(2μl的100μm)和mncl2(1μl的40mm)。
[1036]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰实验室)。
[1037]
5.将反应物在65℃培育20分钟。
[1038]
6.将反应物通过加入tbe-尿素样品缓冲液(novex)停止。
[1039]
7.将反应物在聚丙烯酰胺凝胶(15%)上用tbe缓冲液分离,并通过chemidoc mp成像系统(biorad)进行可视化。
[1040]
凝胶电泳和dna可视化:
[1041]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[1042]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1043]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1044]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[1045]
结果和结论
[1046]
关于使用终止子ix dna聚合酶评估3-o-叠氮基甲基-dttp合并的温度,
[1047]
结果表明在有辅助链连接的情况下3'-o-叠氮基甲基-dttp合并达到90%5分钟后。在37℃和47℃下20分钟后,10%的引物保持未延伸。
[1048]
终止子ix dna聚合酶在2mm的mn
2+
离子和37℃的温度下提供了在辅助链的存在下与dna中互补碱基相对地高效率(从前一个循环开始的连接步骤)合并3
’-
o-修饰的dntp的良好条件。
[1049]
步骤2:裂解
[1050]
第二步描述了用核酸内切酶v一步裂解多核苷酸(图19a)。
[1051]
材料和方法
[1052]
材料
[1053]
1.实例3中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图19d的序列表)。
[1054]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1055]
方法
[1056]
使用以下程序进行寡核苷酸的裂解反应:
[1057]
1.用移液管(gilson)将41μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[1058]
2.然后将5μl的10x reactionneb(50mm的醋酸钾、20mm的tris-乙酸盐、10mm的醋酸镁、1mm的dtt,ph值7.9在25℃下)加入到相同的eppendorf管中。
[1059]
3.将1μl每种寡核苷酸(图19d);模板(seq id no:31)或任何荧光标记的长寡链,具有t的引物(seq id no:32)和对照(seq id no:33)以及辅助链(seq id no:34),均为5pmol加入到同一试管中。
[1060]
4.将1μl人类核酸内切酶v(endo v)neb(10单位/μl)加入到同一试管中。
[1061]
5.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1062]
6.通常在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1063]
使用下面概述的方案纯化样品混合物:
[1064]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1065]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1066]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph 7.5和80%乙醇),并以6000rpm离心1分钟。
[1067]
4.将流过液丢弃并将旋转柱以13000rpm再离心1分钟以去除残余的pe缓冲液。
[1068]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1069]
6.对于dna洗脱,将50μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)加入柱膜中心并在室温下静置1分钟。
[1070]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[1071]
使用以下方案测定纯化的dna浓度:
[1072]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1073]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1074]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1075]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。凝胶电泳和dna可视化:
[1076]
1.将5μl的dna和tbe-尿素样品缓冲液(novex)加入无菌1.5ml eppendorf管中,并使用加热热块(eppendorf)加热至95℃,持续2分钟。
[1077]
2.然后dna混合物加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris、89mm硼酸和2mm edta)。
[1078]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1079]
4.采用使用cy3 leds的chemidoc mp(biorad)进行检测和dna在凝胶中的可视化。可视化和分析在image lab 2.0平台上进行。
[1080]
结果和结论
[1081]
来自实施例3的裂解结果显示,在存在或不存在辅助链的情况下,核酸内切酶v可以获得显著高的裂解dna产量(图19c)。
[1082]
步骤3:连接
[1083]
第三步描述了在辅助链存在下用dna连接酶连接多核苷酸。图20a中示出了图解说明。
[1084]
材料和方法
[1085]
材料
[1086]
1.实例3中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图20b的序列表)。
[1087]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1088]
方法
[1089]
使用以下程序进行寡核苷酸的连接反应:
[1090]
1.用移液管(gilson)将16μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[1091]
2.然后将10μl的2x快速连接反应缓冲液neb(132mm tris-hcl、20mm mgcl2、2mm二硫苏糖醇、2mm atp,15%聚乙二醇(peg6000)和ph 7.6,在25℃)加入到相同的eppendorf管中。
[1092]
3.将1μl每种寡核苷酸(图20b);tamra或任何荧光标记的磷酸酯链(seq id no:35),具有t(seq id no:36)的引物和肌苷链(seq id no:37)和辅助链(seq id no:38),均为5pmol加入到同一试管中。
[1093]
4.将1μl的quick t4 dna连接酶neb(400单位/μl)加入同一试管中。
[1094]
5.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[1095]
6.通常在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[1096]
7.使用如上所述的纯化步骤1-7中概述的方案纯化反应混合物。
[1097]
使用以下方案测定纯化的dna浓度:
[1098]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1099]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1100]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1101]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[1102]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照上述步骤5-8中的程序可视化。没有引入条件或试剂的变化。
[1103]
凝胶电泳和dna可视化:
[1104]
1.将5μl的dna和tbe-尿素样品缓冲液(novex)加入无菌1.5ml eppendorf管中,并使用加热热块(eppendorf)加热到95℃,持续2分钟。
[1105]
2.然后dna混合物加载到15%tbe-尿素凝胶1.0mm x 10孔(invitrogen)的孔中,所述凝胶含有预热的1x tbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1106]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1107]
4.采用使用cy3 leds的chemidoc mp(biorad)进行检测和dna在凝胶中的可视化。可视化和分析在image lab 2.0平台上进行。
[1108]
步骤4:脱保护
[1109]
在dna模型上研究了脱保护步骤(图21a),该模型带有由通过终止子ix dna聚合酶合并3'-o-叠氮甲基-dntp引入到dna的3
′-
o-叠氮甲基基团。通过三(羧乙基)膦(tcep)进行脱保护,并且当将所有天然dntp的混合物加入到纯化的脱保护dna的溶液中时通过延伸反
应进行监测。
[1110]
材料和方法
[1111]
材料
[1112]
1.实例3中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图21i序列)。
[1113]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1114]
3.酶购自新英格兰实验室。
[1115]
方法
[1116]
1.将2μl的10x缓冲液(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4、0.1%x-100,ph 8.8,新英格兰实验室)与12.25μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1117]
2.将1μl的10μm引物(10pmol,1当量)(seq id no:39,图21i)和1.5μl的10μm模板-a/g/t/c(15pmol,1.5当量)(seq id no:40-43,图21i)加入反应混合物中。
[1118]
3.加入3
’-
o-修饰的-dttp/dctp/datp/dgtp(2μl的100μm)和mncl2(1μl的40mm)。
[1119]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰实验室)。
[1120]
5.将反应物在37℃培育5分钟。
[1121]
6.取出4μl样品并与0.5μl的5mm dntp混合物混合并使其反应10分钟以进行对照反应。
[1122]
7.将在1m tris缓冲液ph 7.4中的40μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1123]
8.通过用20μl的缓冲液洗脱,使用qiagen核苷酸去除试剂盒纯化反应混合物。
[1124]
9.将1μl的5mm dntp混合物和1μl的deepvent(exo-)dna聚合酶加入到纯化的反应混合物中并使其反应10分钟。
[1125]
10.将反应物通过加入tbe-尿素样品缓冲液(novex)停止。
[1126]
11.将反应物在聚丙烯酰胺凝胶(15%)上用tbe缓冲液分离,并通过chemidoc mp成像系统(biorad)进行可视化。
[1127]
结果和结论
[1128]
50mm tcep不足以在0.2μm dna模型上高效裂解3'-o-叠氮甲基(图21h)。相反,300mm tcep成功地在0.2μm dna模型上以95%的效率裂解3'-o-叠氮甲基(图21h)。
[1129]
实例4有双发夹模型的版本2化学法.
[1130]
该实施例描述了使用4个步骤在双发夹模型上合成多核苷酸:从切口位点合并3
’-
o-修饰的dntp;在第一步在与天然互补核苷酸相对地发生的情况下裂解、连接和脱保护,该天然互补核苷酸位于支持链中与通用核苷酸相邻,在该特定情况下通用核苷酸为肌苷。
[1131]
步骤1:合并
[1132]
第一步描述了通过dna聚合酶的酶促合并将3
’-
o-保护的单核苷酸受控的添加到寡核苷酸中(图22a)。
[1133]
材料和方法
[1134]
材料
[1135]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis。columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1136]
2.寡核苷酸是内部设计的并从sigma-aldrich获得(图22c)。以100μm的浓度制备储备溶液。
[1137]
3.使用终止子ix dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。
[1138]
测试3'-o-叠氮甲基-dttp的合并:
[1139]
3'-o-叠氮甲基-dttp:
[1140][1141]
方法
[1142]
1.2μl缓冲液(20mm的tris-hcl,10mm的(nh4)2so4,10mm kcl,2mm mgso4、0.1%x-100,ph 8.8,新英格兰实验室)与10.25μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1143]
2.将0.5μl的10μm发夹寡核苷酸(5pmol,1当量)(seq id no:44,图22c)加入到反应混合物中。
[1144]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[1145]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰生物实验室)。
[1146]
5.将反应物在65℃培育20分钟。
[1147]
6.将反应物通过加入tbe-尿素样品缓冲液(novex)停止。
[1148]
7.将反应物在聚丙烯酰胺凝胶(15%)上用tbe缓冲液分离,并通过chemidoc mp成像系统(biorad)进行可视化。
[1149]
凝胶电泳和dna可视化:
[1150]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[1151]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1152]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1153]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[1154]
结果
[1155]
dna聚合酶在发夹构建体中合并与其天然互补碱基相对的3
’-
o-修饰的dttp。
[1156]
步骤2:裂解
[1157]
第二步描述了用核酸内切酶v在此特定情况下一步裂解发夹模型(图23a)。
[1158]
材料和方法
[1159]
材料
[1160]
1.实例4中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图23c序列)。
[1161]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1162]
方法
[1163]
发夹寡核苷酸的裂解反应使用以下步骤进行:
[1164]
1.用移液管(gilson)将43μl无菌蒸馏水(elga veolia)转移到1.5ml eppendorf管中。
[1165]
2.然后将5μl的10x reactionneb(50mm的醋酸钾,20mm的tris-乙酸盐,10mm的醋酸镁,1mm的dtt,ph值7.9在25℃下)加入到相同的eppendorf管中。
[1166]
3.将具有5pmol量的1μl发夹寡核苷酸(seq id no:45,图23c)加入同一试管中。
[1167]
4.将1μl人类核酸内切酶v(endo v)neb(30单位/μl)加入到同一试管中。
[1168]
5.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1169]
6.通常在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1170]
使用下面概述的方案纯化样品混合物:
[1171]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1172]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1173]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph 7.5和80%乙醇),并以6000rpm离心1分钟。
[1174]
4.将流过液丢弃并将旋转柱以13000rpm离心另外的1分钟以去除残余的pe缓冲液。
[1175]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1176]
6.对于dna洗脱,将50μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)加入柱膜中心并在室温下静置1分钟。
[1177]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[1178]
使用以下方案测定纯化的dna浓度:
[1179]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1180]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1181]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1182]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[1183]
凝胶电泳和dna可视化:
[1184]
1.将5μl的dna和tbe-尿素样品缓冲液(novex)加入无菌1.5ml eppendorf管中,并使用加热thermomixer(eppendorf)加热至95℃,持续2分钟。
[1185]
2.然后dna混合物加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1186]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1187]
4.采用使用cy3 leds的chemidoc mp(biorad)进行检测和dna在凝胶中的可视化。可视化和分析在image lab 2.0平台上进行。
[1188]
结果和结论
[1189]
来自实施例4的裂解结果显示,使用核酸内切酶v在37℃下获得了显著高的消化的发夹dna产率(图23b)。
[1190]
步骤3:连接
[1191]
第三步描述了用dna连接酶连接发夹模型。示意图如图24a所示。
[1192]
材料和方法
[1193]
材料
[1194]
1.实例4中使用的寡核苷酸是内部设计的并由sigma aldrich合成(参见图24c序列)。
[1195]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1196]
方法
[1197]
使用以下程序进行寡核苷酸的连接反应:
[1198]
1.使用移液管(gilson)将1μl(5pmol)tamra或任何荧光标记磷酸发夹寡核苷酸(seq id no:46)转移入1.5毫升eppendorf管中。
[1199]
2.然后将15μl(100pmol)含肌苷的发夹构建体(seq id no:47)加入到相同的管中,并通过用移液管重悬浮轻轻混合3秒。
[1200]
3.将40μl的blunt/ta dna连接酶neb(180单位/μl)加入同一试管中。
[1201]
4.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[1202]
5.通常在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[1203]
6.使用上面的纯化步骤1-7中概述的方案纯化反应混合物。
[1204]
使用以下方案测定纯化的dna浓度:
[1205]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1206]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1207]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1208]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[1209]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照上述步骤5-8中的方法可视化。没有引入条件或试剂的变化。
[1210]
凝胶电泳和dna可视化。
[1211]
1.将5μl的dna和tbe-尿素样品缓冲液(novex)加入无菌1.5ml eppendorf管中,并使用加热thermomixer(eppendorf)加热至95℃,持续2分钟。
[1212]
2.然后dna混合物加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1213]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1214]
4.采用使用cy3 leds的chemidoc mp(biorad)进行检测和dna在凝胶中的可视化。可视化和分析在image lab 2.0平台上进行。
[1215]
结果
[1216]
在辅助链的存在下,在室温(24℃)下用发夹/ta dna连接酶连接发夹寡核苷酸,得到高产率的连接产物。1分钟后连接的发夹寡核苷酸显示出高产率的连接dna产物,比率为约85%。2分钟后连接的发夹寡核苷酸显示出高产率的连接dna,比率为约85%。3分钟后连接的发夹寡核苷酸显示出高产率的连接dna产物,比率为约85%。4分钟后连接的发夹寡核苷酸显示高产率的连接dna产物,比率为约>85%(图24b)。
[1217]
实例5.版本2化学方法-双发夹模型的完整循环.
[1218]
该实施例描述了使用4个步骤在双发夹模型上合成多核苷酸:从切口位点合并3
’-
o-修饰的dntp;在第一步在与天然互补核苷酸相对地发生的情况下裂解、连接和脱保护,该天然互补核苷酸位于支持链中与通用核苷酸相邻,在该特定情况下通用核苷酸为肌苷。发夹的一端用作附着锚。
[1219]
该方法首先是通过dna聚合酶的酶促合并,然后肌苷裂解、连接和脱保护,将3'-o-保护的单核苷酸受控的添加到寡核苷酸中(图25a)。
[1220]
材料和方法
[1221]
材料
[1222]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis。columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1223]
2.寡核苷酸是内部设计的并从sigma-aldrich获得(图25c)。以100μm的浓度制备储备溶液。
[1224]
3.使用终止子ix dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。
[1225]
测试3'-o-叠氮甲基-dttp的合并:
[1226]
3'-o-叠氮甲基-dttp:
[1227][1228]
方法
[1229]
1.将2μl的10x缓冲液(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4、0.1%x-100,ph 8.8,新英格兰生物实验室(new england biolabs))与12.5μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1230]
2.将2μl的10μm双发夹模型寡核苷酸(20pmol,1当量)(seq id no:48,图25c)加入到反应混合物中。
[1231]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[1232]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰生物实验室)。
[1233]
5.将反应物在37℃培育10分钟。
[1234]
6.从反应混合物取出等分试样(5μl),以及加入0.5μl的天然dntp混合物并使其反应10分钟。通过凝胶电泳分析反应。
[1235]
7.使用纯化步骤1-7中概述的方案纯化反应混合物。
[1236]
8.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1237]
9.将1μl人类核酸内切酶v(endo v)neb(30单位/μl)加入到同一试管中。
[1238]
10.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1239]
11.在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1240]
12.将等分试样(5μl)从反应混合物取出,并在使用tbe缓冲液的聚丙烯酰胺凝胶(15%)上分析,并用chemidoc mp成像系统(biorad)进行可视化。
[1241]
13.使用上面纯化步骤1-7中概述的方案纯化反应混合物。
[1242]
14.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1243]
15.将10μl用于连接的100μm链(1nmol)(seq id no:49,图25c)加入到反应混合物中。
[1244]
16.将40μl的blunt/ta dna连接酶neb(180单位/μl)加入到纯化的dna样品中。
[1245]
17.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[1246]
18.将在1m tris缓冲液ph 7.4中的40μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1247]
19.通过用20μl的缓冲液洗脱,使用qiagen核苷酸去除试剂盒纯化反应混合物。
[1248]
凝胶电泳和dna可视化:
[1249]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[1250]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mm x 10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1251]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1252]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[1253]
使用以下方案测定纯化的dna浓度:
[1254]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1255]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1256]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1257]
4.通过将2μl样品加到基座上并在触摸屏上选择测量图标来测量dna浓度。
[1258]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照章节2步骤5-8中的程序可视化。没有引入条件或试剂的变化。
[1259]
使用下面概述的方案在每个步骤后纯化样品混合物:
[1260]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1261]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1262]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph7.5和80%乙醇),并以6000rpm离心1分钟。
[1263]
4.将流过液丢弃并将旋转柱以13000rpm离心另外的1分钟以去除残余的pe缓冲液。
[1264]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1265]
6.对于dna洗脱,将20μl适当的反应缓冲液加入到柱膜中心,并在室温下静置1分钟。
[1266]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[1267]
结果
[1268]
dna聚合酶在双发夹构建体中合并与其天然互补碱基相对的3'-o-修饰的dttp(图25b)。
[1269]
实例6.版本2化学法-使用辅助链的单发夹模型的完整循环.
[1270]
该实施例描述了使用4个步骤在单发夹模型上合成多核苷酸:从切口位点合并3
’-
o-修饰的dntp;在第一步在与天然互补碱基相对地发生的情况下裂解、连接和脱保护。dna合成在所述过程中使用辅助链。
[1271]
该方法首先是通过dna聚合酶的酶促合并,然后肌苷裂解、连接和脱保护,将3'-o-保护的单核苷酸受控的添加到寡核苷酸中(图26a)。
[1272]
材料和方法
[1273]
材料
[1274]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis。columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1275]
2.寡核苷酸是内部设计的并从sigma aldrich获得(图26b)。以100μm的浓度制备储备溶液。
[1276]
3.使用终止子ix dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。
[1277]
测试3'-o-叠氮甲基-dttp的合并:
[1278]
3'-o-叠氮甲基-dttp:
[1279][1280]
方法
[1281]
1.2μl缓冲液(20mm的tris-hcl,10mm的(nh4)2so4,10mm kcl,2mm mgso4、0.1%x-100,ph 8.8,新英格兰实验室)与12.5μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1282]
2.将2μl的10μm单发夹模型寡核苷酸(20pmol,1当量)(seq id no:50,图26b)和辅助链(30pmol,1.5当量)(seq id no:51,图26b)加入到反应混合物中。
[1283]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[1284]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰生物实验室)。
[1285]
5.将反应物在37℃培育10分钟。
[1286]
6.从反应混合物取出等分试样(5μl),以及加入0.5μl的天然dntp混合物并使其反应10分钟。通过凝胶电泳分析反应。
[1287]
7.使用上面纯化步骤1-7中概述的方案纯化反应混合物。
[1288]
8.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1289]
9.将1μl人类核酸内切酶v(endo v)neb(30单位/μl)加入到同一试管中。
[1290]
10.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1291]
11.在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1292]
12.将等分试样(5μl)从反应混合物取出,并在使用tbe缓冲液的聚丙烯酰胺凝胶
(15%)上分析,并用chemidoc mp成像系统(biorad)进行可视化。
[1293]
13.使用上面纯化步骤1-7中概述的方案纯化反应混合物。
[1294]
14.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1295]
15.将用于连接的10μl的100μm链(1 nmol)(seq id no:52,图26b)和用于连接的10μl的100μm辅助链(1nmol)(seq id no:53,图26b)加入到反应混合物中。
[1296]
16.将40μl的blunt/ta dna连接酶neb(180单位/μl)加入同一试管中。
[1297]
17.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[1298]
18.将在1m tris缓冲液ph 7.4中的40μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1299]
19.通过用20μl的1xneb缓冲液洗脱,使用qiagen核苷酸去除试剂盒纯化反应混合物。
[1300]
20.通常在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[1301]
凝胶电泳和dna可视化:
[1302]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[1303]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mm x 10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1304]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1305]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[1306]
使用以下方案测定纯化的dna浓度:
[1307]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1308]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1309]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1310]
4.通过将2μl样品加在基座上并在触摸屏上选择测量图标来测量dna浓度。
[1311]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照上述步骤5-8中的程序可视化。没有引入条件或试剂的变化。
[1312]
使用下面概述的方案在每个步骤后纯化样品混合物:
[1313]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1314]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1315]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph 7.5和80%乙醇),并以6000rpm离心1分钟。
[1316]
4.将流过液丢弃并将旋转柱以13000rpm离心另外的1分钟以去除残余的pe缓冲
液。
[1317]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1318]
6.对于dna洗脱,将20μl适当的反应缓冲液加入到柱膜中心,并在室温下静置1分钟。
[1319]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[1320]
实例7.版本3化学-双发夹模型的完整循环.
[1321]
该实施例描述了使用4个步骤在双发夹构建体模型上合成多核苷酸:从切口位点合并3
’-
o-修饰的dntp;在第一步在与通用核苷酸相邻(在该特定情况下是苷酸)相对地发生的情况下裂解、连接和脱保护。
[1322]
该方法首先是通过dna聚合酶的酶促合并,然后肌苷裂解、连接和脱保护,将3'-o-保护的单核苷酸受控的添加到寡核苷酸中(图27a)。
[1323]
材料和方法
[1324]
材料
[1325]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis.columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1326]
2.寡核苷酸是内部设计的并从sigma-aldrich获得(图27b)。以100μm的浓度制备储备溶液。
[1327]
3.其由new england biolabs工程化的终止子ix dna聚合酶具有增强的合并3-o-修饰的dntp的能力。
[1328]
测试3'-o-叠氮甲基-dttp的合并:
[1329]
3'-o-叠氮甲基-dttp:
[1330][1331]
方法
[1332]
1.将2μl的10x缓冲液(20mm tris-hcl、10mm(nh4)2so4、10mm kcl、2mm mgso4、0.1%x-100,ph 8.8,新英格兰生物实验室(new england biolabs))与12.5μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1333]
2.将2μl的10μm双发夹模型寡核苷酸(20pmol,1当量)(seq id no:54,图27b)加入到反应混合物中。
[1334]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[1335]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰生物实验室)。
[1336]
5.将反应物在37℃培育10分钟。
[1337]
6.从反应混合物取出等分试样(5μl),以及加入0.5μl的天然dntp混合物并使其反应10分钟。通过凝胶电泳分析反应。
[1338]
7.使用纯化步骤1-7中概述的方案纯化反应混合物。
[1339]
8.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1340]
9.将1μl人类核酸内切酶v(endo v)neb(30单位/μl)加入到同一试管中。
[1341]
10.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1342]
11.在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1343]
12.将等分试样(5μl)从反应混合物取出,并在使用tbe缓冲液的聚丙烯酰胺凝胶(15%)上分析,并用chemidoc mp成像系统(biorad)进行可视化。
[1344]
13.使用上面纯化步骤1-7中概述的方案纯化反应混合物。
[1345]
14.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1346]
15.将10μl用于连接的100μm链(1nmol)(seq id no:55,图27b)加入到反应混合物中。
[1347]
16.将40μl的blunt/ta dna连接酶neb(180单位/μl)加入同一试管中。
[1348]
17.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育20分钟。
[1349]
18.将在1m tris缓冲液ph 7.4中的40μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1350]
19.通过用20μl的1xneb缓冲液洗脱,使用qiagen核苷酸去除试剂盒纯化反应混合物。
[1351]
20.通常在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[1352]
凝胶电泳和dna可视化:
[1353]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[1354]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mm x 10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1355]
3.将x-cell sure lock模块(novex)固定到位并在以下条件下进行电泳;260v,90amp,室温下40分钟。
[1356]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[1357]
使用以下方案测定纯化的dna浓度:
[1358]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1359]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1360]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复第2步。
[1361]
4.通过将2μl样品加在基座上并在触摸屏上选择测量图标来测量dna浓度。
[1362]
5.将纯化的dna在聚丙烯酰胺凝胶上运行,并按照章节2步骤5-8中的程序可视化。没有引入条件或试剂的变化。
[1363]
使用下面概述的方案在每个步骤后纯化样品混合物:
[1364]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1365]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1366]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph7.5和80%乙醇),并以6000rpm离心1分钟。
[1367]
4.将流过液丢弃并将旋转柱以13000rpm离心另外的1分钟以去除残余的pe缓冲液。
[1368]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1369]
6.对于dna洗脱,将20μl适当的反应缓冲液加入到柱膜中心,并在室温下静置1分钟。
[1370]
7.然后将管以13000rpm离心1分钟。测量洗脱的dna浓度并储存在-20℃下以备后用。
[1371]
实例8.版本2化学-双发夹模型的完整双循环实验.
[1372]
该实施例描述了在双发夹模型上使用4个步骤合成多核苷酸的完整双循环实验:从切口位点合并3'-o-修饰的dntp;在第一步在与互补碱基相对地发生的情况下脱保护、裂解和连接。
[1373]
该方法首先是通过dna聚合酶的酶促合并,然后脱保护、肌苷裂解和连接,将3'-o-保护的单核苷酸受控地加入寡核苷酸,如图28a所示的第一个循环的反应示意图所示。图28b显示了第二循环的反应示意图。
[1374]
材料和方法
[1375]
材料
[1376]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis.columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1377]
2.寡核苷酸是内部设计的并从sigma-aldrich获得(图28d)。以100μm的浓度制备储备溶液。
[1378]
3.其由new england biolabs工程化的终止子ix dna聚合酶具有增强的合并3
’-
o-修饰的dntp的能力。
[1379]3’-
o-叠氮甲基-dttp和3'-o-叠氮甲基-dctp被用于合并:
[1380][1381]
方法
[1382]
第1循环:
[1383]
1.将2μl的缓冲液(20mm tris-hcl,10mm(nh4)2so4,10mm kcl,2mm mgso4,0.1%x-100,ph 8.8,新英格兰实验室)与12.5μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1384]
2.将2μl的10μm双发夹模型寡核苷酸(20pmol,1当量)(seq id no:56,图28d)加入到反应混合物中。
[1385]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(1μl的40mm)。
[1386]
4.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰实验室)。
[1387]
5.将反应物在37℃培育10分钟。
[1388]
6.从反应混合物取出等分试样(5μl),以及加入0.5μl的天然dntp混合物并使其反应10分钟。通过凝胶电泳分析反应。
[1389]
7.将在1m tris缓冲液ph=7.4中的40μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1390]
8.使用纯化步骤1-7中概述的方案纯化反应混合物。
[1391]
9.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1392]
10.将1μl人类核酸内切酶v(endo v)neb(30单位/μl)加入到同一试管中。
[1393]
11.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1394]
12.在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1395]
13.将等分试样(5μl)从反应混合物取出,并在使用tbe缓冲液的聚丙烯酰胺凝胶(15%)上分析,并用chemidoc mp成像系统(biorad)进行可视化。
[1396]
14.使用qiagen核苷酸去除试剂盒,使用纯化步骤1-7中概述的方案纯化反应混合物。
[1397]
15.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1398]
16.将10μl用于连接的100μm链(1nmol)(seq id no:57,图28d)加入到反应混合物中。
[1399]
17.将40μl的blunt/ta dna连接酶neb(180单位/μl)加入同一试管中。
[1400]
18.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在
室温培育20分钟。
[1401]
19.使用纯化步骤1-5中概述的方案,通过链酶抗生物素蛋白磁珠试剂盒纯化反应混合物。
[1402]
20.未连接的寡核苷酸用λ核酸外切酶消化。
[1403]
21.使用qiagen核苷酸去除试剂盒,使用纯化步骤1-7中概述的方案纯化反应混合物。
[1404]
22.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。第2循环:
[1405]
23.加入3
’-
o-修饰的-dctp(2μl的100μm)和mncl2(1μl的40mm)。
[1406]
24.然后加入1.5μl的therminator ix dna聚合酶(15u,新英格兰实验室)。
[1407]
25.将反应物在37℃培育10分钟。
[1408]
26.从反应混合物取出等分试样(5μl),以及加入0.5μl的天然dntp混合物并使其反应10分钟。通过凝胶电泳分析反应。
[1409]
27.将在1m tris缓冲液ph=7.4中的40μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1410]
28.使用纯化步骤1-7中概述的方案纯化反应混合物。
[1411]
29.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1412]
30.将1μl人类核酸内切酶v(endo v)neb(30单位/μl)加入到同一试管中。
[1413]
31.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在37℃培育1小时。
[1414]
32.在培育时间过去之后,将反应物通过酶促热失活(即65℃下20分钟)终止。
[1415]
33.将等分试样(5μl)从反应混合物取出,并在使用tbe缓冲液的聚丙烯酰胺凝胶(15%)上分析,并用chemidoc mp成像系统(biorad)进行可视化。
[1416]
34.使用纯化步骤1-7中概述的方案纯化反应混合物。
[1417]
35.通过20μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1418]
36.将10μl用于连接的100μm链(1nmol)(seq id no:58,图28d)加入到反应混合物中。
[1419]
37.将40μl的blunt/ta dna连接酶neb(180单位/μl)加入同一试管中。
[1420]
38.然后将反应混合物通过用移液管重悬温和混合,13,000rpm下离心5秒钟并在室温培育10分钟。
[1421]
39.在培育时间过去后,加入tbe-尿素样品缓冲液(novex)终止反应。
[1422]
凝胶电泳和dna可视化:
[1423]
1.在无菌1.5ml eppendorf管中将5μl反应混合物加入5μl的tbe-尿素样品缓冲液(novex)中,并使用加热thermomixer(eppendorf)加热至95℃,持续5分钟。
[1424]
2.然后将5μl样品加载到15%tbe-尿素凝胶1.0mmx10孔(invitrogen)的孔中,所述凝胶含有预热的1xtbe缓冲液thermo scientific(89mm tris,89mm硼酸和2mm edta)。
[1425]
3.将x-cell sure lock模块(novex)固定到位并通过应用以下条件下进行电泳;
260v,90amp,室温下40分钟。
[1426]
4.将凝胶通过使用cy3 leds的chemidoc mp(biorad)可视化。可视化和分析在image lab 2.0平台上进行。
[1427]
使用以下方案测定纯化的dna浓度:
[1428]
1.加入2μl灭菌蒸馏水(elga veolia)到基座平衡nanodrop one(thermo scientific)。
[1429]
2.平衡后,用不起毛的镜片清洁纸(whatman)轻轻擦去水。
[1430]
3.通过加入2μl缓冲液eb qiagen(10mm tris.cl,ph 8.5)掩蔽(blank)nanodrop one。然后在掩蔽后重复步骤2。
[1431]
4.通过将2μl样品加在基座上并在触摸屏上选择测量图标来测量dna浓度。
[1432]
使用下面概述的方案,通过qiagen核苷酸去除试剂盒纯化样品混合物:
[1433]
1.将500μl缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1434]
2.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1435]
3.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph7.5和80%乙醇),并以6000rpm离心1分钟。
[1436]
4.将流过液丢弃并将旋转柱以13000rpm再离心1分钟以去除残余的pe缓冲液。
[1437]
5.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1438]
6.对于dna洗脱,将20μl适当的反应缓冲液加入到柱膜中心,并在室温下静置1分钟。
[1439]
7.然后将管以13000rpm离心1分钟。
[1440]
在连接步骤后,使用链酶抗生物素蛋白磁珠通过下面概述的方案纯化样品混合物:
[1441]
1.用200μl结合缓冲液(20mm tris,500mm nacl,ph=7.4)洗涤100μl链酶抗生物素蛋白磁珠(新英格兰实验室)3次。
[1442]
2.连接步骤之后反应混合物与10倍体积的结合缓冲液(20mm的tris,500mm的nacl,ph值=7.4)混合,并用链酶抗生物素蛋白磁珠在20℃培育15分钟。
[1443]
3.用200μl结合缓冲液(20mm tris,500mm nacl,ph=7.4)洗涤链酶抗生物素蛋白磁珠3次。
[1444]
4.用去离子水洗涤链酶抗生物素蛋白磁珠3次。
[1445]
5.通过加热至95℃持续3分钟将寡核苷酸用40μl去离子水洗脱。
[1446]
图28c中所示的结果证明了使用本发明的示例性方法的两个完合并成循环的性能。
[1447]
实例9.版本2化学-单发夹模型上的完整三循环实验。
[1448]
该实施例描述了在双发夹模型上使用5个步骤合成多核苷酸的完整三循环实验:从切口位点合并3'-o-修饰的dntp;在第一步在与互补碱基相对地发生的情况下脱保护、裂解、连接和变性步骤。
[1449]
所述方法的示例性示意图概述显示在图33、34和35中。
[1450]
该方法首先是通过dna聚合酶的酶促合并,然后肌苷脱保护、裂解、连接和辅助链
的变性,将3'-o-保护的单核苷酸受控的添加到寡核苷酸中。图33显示了涉及酶合并、脱保护、裂解、连接和变性步骤的第1个完整循环。在所述实施例中,寡核苷酸由t核苷酸延伸。图34显示了第1个循环后的第2个完整循环,包括酶促合并、脱保护、裂解、连接步骤和变性步骤。在所述实施例中,寡核苷酸由t核苷酸延伸。图35显示了第2个循环后的第3个完整循环,包括酶促合并、脱保护、裂解、连接和变性步骤。在所述实施例中,寡核苷酸由t核苷酸延伸。
[1451]
材料和方法
[1452]
材料
[1453]
1.根据描述于以下的方案内部合成3'-o-经修饰的dntp:博士毕业论文jian wu:molecular engineering of novel nucleotide analogues for dna sequencing by synthesis,columbia university,2008.用于合成的方案还描述于专利申请公开:j.william efcavitch,juliesta e.sylvester,modified template-independent enzymes for polydeoxynucleotide synthesis,molecular assemblies us2016/0108382a1。
[1454]
2.寡核苷酸是内部设计的并从integrated dna technologies,sigma-aldrich获得(图36)。以100μm的浓度制备储备溶液。
[1455]
3.使用终止子x dna聚合酶,其由new england biolabs工程化,具有增强的合并3-o-修饰的dntp的能力。可以替代地使用任何可以合并修饰的dntp的dna聚合酶或其他酶。
[1456]
使用3'-o-叠氮甲基-dttp合并:
[1457][1458]
方法
[1459]
第1循环:
[1460]
1.将20μl的10x缓冲液(20mm tris-hcl,10mm(nh4)2so4,10mm kcl,2mm mgso4,0.1%x-100,ph 8.8,新英格兰实验室)和mncl2溶液(10μl的40mm)与139μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1461]
2.将20μl的100μm单发夹模型寡核苷酸(2nmol,1当量)(seq id no:59,图36)加入到反应混合物中。
[1462]
3.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1463]
4.加入3'-o-修饰的-dttp(10μl,2mm)。
[1464]
5.然后加入5μl的终止子ix dna聚合酶(50u,new england biolabs)。然而,可以使用任何可以合并修饰的dntp的dna聚合酶或其他酶。
[1465]
6.将反应物在37℃培育30分钟。
[1466]
7.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒纯化反应混合物。
[1467]
8.通过200μl的te缓冲液将dna样品洗脱到干净的eppendorf管中。
[1468]
9.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1469]
10.将400μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1470]
11.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒纯化反应混合物。
[1471]
12.通过150μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph 7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1472]
13.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1473]
14.将5μl的人类核酸内切酶v(endo v)neb(30单位/μl)加入洗脱液并在37℃培育30分钟。可以使用任何合适的替代性核酸内切酶。
[1474]
15.在培育时间过去之后,将反应物通过在65℃下酶促热失活20分钟终止。
[1475]
16.将等分试样(5μl)从反应混合物取出,并在聚丙烯酰胺凝胶上分析。
[1476]
17.通过qiagen核苷酸去除试剂盒,使用纯化步骤66-72中概述的方案纯化反应混合物。
[1477]
18.通过100μl的t3 dna连接酶缓冲液(2x浓度)将dna样品洗脱到干净的eppendorf管中。
[1478]
19.将20μl的100μm用于连接的肌苷链(2nmol)和20μl的100μm用于连接的辅助链(2nmol)(seq id no:60,51,图36)以及40μl水加入到反应混合物中。
[1479]
20.将20μl的t3 dna连接酶neb(3000单位/μl)加入同一试管中(这可包括任何dna连接酶)并在室温下培育30分钟。
[1480]
使用对于链酶抗生物素蛋白磁珠试剂盒的方案纯化反应混合物,包括纯化步骤73-78中概述的变性步骤。
[1481]
21.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒的方案纯化反应混合物。
[1482]
22.通过100μl的te缓冲液将dna样品洗脱到干净的eppendorf管中。
[1483]
第2循环:
[1484]
23.加入15μl的10x缓冲液(20mm tris-hcl,10mm(nh4)2so4,10mm kcl,2mm mgso4,0.1%x-100,ph 8.8,new england biolabs)、mncl2溶液(7.5μl,40mm)和19μl去离子水。
[1485]
24.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1486]
25.加入3'-o-修饰的-dttp(7.5μl,2mm)。
[1487]
26.然后加入5μl的终止子x dna聚合酶(50u,new england biolabs)。可以使用任何可以合并修饰的dntp的dna聚合酶。
[1488]
27.将反应物在37℃培育30分钟。
[1489]
28.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒纯化反应混合物。
[1490]
29.通过100μl的te缓冲液将dna样品洗脱到干净的eppendorf管中。
[1491]
30.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1492]
31.将200μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1493]
32.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒纯化反应混合物。
[1494]
33.通过100μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1495]
34.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1496]
35.将5μl的人类核酸内切酶v(endo v)neb(30单位/μl)加入洗脱液并在37℃培育30分钟。可以使用任何合适的替代性核酸内切酶。
[1497]
36.在培育时间过去之后,将反应物通过在65℃下酶促热失活20分钟终止。
[1498]
37.将等分试样(5μl)取出反应混合物,并在聚丙烯酰胺凝胶上分析。
[1499]
38.通过qiagen核苷酸去除试剂盒,使用纯化步骤66-72中概述的方案纯化反应混合物。
[1500]
39.通过60μl的t3 dna连接酶缓冲液(2x浓度)将dna样品洗脱到干净的eppendorf管中。
[1501]
40.将20μl的100μm用于连接的肌苷链(2nmol)和20μl的100μm用于连接的辅助链(2nmol)(seq id no:60,51,图36)以及10μl去离子水加入到反应混合物中。
[1502]
41.将10μl的t3 dna连接酶neb(3000单位/μl)加入同一试管中并在室温下培育30分钟。可以使用任何合适的dna连接酶。
[1503]
42.使用对于链酶抗生物素蛋白磁珠试剂盒的方案纯化反应混合物,包括纯化步骤73-78中概述的变性步骤。
[1504]
43.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒的方案纯化反应混合物。
[1505]
44.通过46μl的te缓冲液将dna样品洗脱到干净的eppendorf管中。
[1506]
第3循环:
[1507]
45.加入6μl的10x缓冲液(20mm tris-hcl,10mm(nh4)2so4,10mm kcl,2mm mgso4,0.1%x-100,ph 8.8,new england biolabs)、mncl2溶液(3μl,40mm)。
[1508]
46.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1509]
47.加入3'-o-修饰的dttp(6μl的200μm)。
[1510]
48.然后加入3μl的therminatorx dna聚合酶(30u,new england biolabs)。可以使用任何可以合并修饰的dntp的dna聚合酶或其他合适的酶。
[1511]
49.将反应物在37℃培育30分钟。
[1512]
50.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒纯化反应混合物。
[1513]
51.通过50μl的te缓冲液将dna样品洗脱到干净的eppendorf管中。
[1514]
52.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1515]
53.将100μl的500mm tcep加入到反应混合物中,并使其在37℃反应10分钟。
[1516]
54.使用在纯化步骤66-72中概述的qiagen核苷酸去除试剂盒纯化反应混合物。
[1517]
55.通过49μl的neb反应(50mm乙酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9,25℃下)将dna样品洗脱到干净的eppendorf管中。
[1518]
56.从反应混合物取出等分试样(4μl),以及加入0.5μl的天然dntp混合物(4mm)和0.5μl的bst dna聚合酶以及0.5μl的sulfolobus dna聚合酶iv并使其反应10分钟。通过凝胶电泳分析反应。
[1519]
57.将5μl的人类核酸内切酶v(endo v)neb(30单位/μl)加入洗脱液并在37℃培育30分钟。或者可以使用任何合适的核酸内切酶。
[1520]
58.在培育时间过去之后,将反应物通过在65℃下酶促热失活20分钟终止。
[1521]
59.将等分试样(5μl)取出反应混合物,并在聚丙烯酰胺凝胶上分析。
[1522]
60.通过qiagen核苷酸去除试剂盒,使用纯化步骤66-72中概述的方案纯化反应混合物。
[1523]
61.通过30μl的t3 dna连接酶缓冲液(2x浓度)将dna样品洗脱到干净的eppendorf管中。
[1524]
62.将10μl的100μm用于连接的肌苷链(2nmol)、10μl的100μm用于连接的辅助链(2nmol)(seq id no:60,51,图36)以及5μl水加入到反应混合物中。
[1525]
63.将5μl的t3 dna连接酶neb(3000单位/μl)加入同一试管中。(这可包含任何dna连接酶)并在室温下培育30分钟。
[1526]
64.通过凝胶电泳分析反应混合物。
[1527]
使用下述方案在合并、解封闭和裂解步骤后通过qiagen核苷酸去除试剂盒纯化反应混合物:
[1528]
65.将10体积的缓冲液pni qiagen(5m氯化胍)加入到样品中并通过用移液管重悬浮温和混合。
[1529]
66.将混合物转移到qiaquick旋转柱(qiagen)并以6000rpm离心1分钟。
[1530]
67.离心后,弃去流过液,向旋转柱中加入750μl缓冲液pe qiagen(10mm tris-hcl ph7.5和80%乙醇),并以6000rpm离心1分钟。
[1531]
68.将流过液丢弃并将旋转柱以13000rpm再离心1分钟以去除残余的pe缓冲液。
[1532]
69.然后将旋转柱置于无菌的1.5ml eppendorf管中。
[1533]
70.对于dna洗脱,将20-200μl适当的反应缓冲液加入到柱膜中心,并在室温下静置1分钟。
[1534]
71.然后将管以13000rpm离心1分钟。
[1535]
通过下面概述的方案,在使用涉及变性步骤的链酶抗生物素蛋白磁珠的连接步骤后进行反应的纯化:
[1536]
72.用100μl结合缓冲液(20mm tris,500mm nacl,ph=7.4)洗涤200μl链酶抗生物
素蛋白磁珠(new england biolabs)3次。
[1537]
73.连接步骤之后反应混合物与10倍体积的结合缓冲液(20mm的tris,500mm的nacl,ph值=7.4)混合,并用链酶抗生物素蛋白磁珠在20℃使其培育15分钟。
[1538]
74.用200μl结合缓冲液(20mm tris,500mm nacl,ph=7.4)洗涤链酶抗生物素蛋白磁珠3次。
[1539]
75.为了去除辅助链,在放置于磁体上的200μl结合缓冲液(20mm tris,500mm nacl,ph=7.4)中将链酶抗生物素蛋白磁珠加热至80℃,并将上清液迅速丢弃。
[1540]
76.用去离子水洗涤链酶抗生物素蛋白磁珠3次。
[1541]
77.通过加热至95℃持续3分钟将寡核苷酸用50-100μl去离子水洗脱。
[1542]
结果和结论
[1543]
图37描绘了显示对应于完整三循环实验的反应产物的凝胶,所述实验包括:合并、解封闭、裂解和连接步骤。所示结果证明了使用本发明的示例性方法的三个完合并成循环的性能。
[1544]
实例10.衍生化聚丙烯酰胺表面并随后固定分子。
[1545]
所述实施例描述了使用n-(5-溴乙酰胺基戊基)丙烯酰胺(brapa)在聚丙烯酰胺表面上呈现溴乙酰基,并且随后通过它们与溴乙酰基的共价偶联来表面固定硫醇化分子。
[1546]
材料和方法
[1547]
通过在丙酮、乙醇和水中依次超声处理,每次10分钟,并用氩气干燥,清洁玻璃显微镜载玻片和盖玻片。将清洁的盖玻片在聚苯乙烯培养皿中用三氯(1h,1h,2h,2h-全氟辛基)硅烷在气相中硅烷化,在乙醇中超声处理两次并用ar干燥(下文
‘
氟化盖玻片')。在玻璃显微镜载玻片上,将4%丙烯酰胺/n,n'-亚甲基双丙烯酰胺(19:1)溶液与100μl的10%(w/v)过硫酸铵(aps)、10μl的四甲基乙二胺(temed)混合,用0、0.1、0.2和0.3%(w/v)的n-(5-溴乙酰胺基戊基)丙烯酰胺(brapa)掺杂,并快速分配到4mm直径的橡胶垫圈中,随后用氟化盖玻片夹在中间,氟化面朝向丙烯酰胺溶液,并聚合10分钟。10分钟后,将表面浸入去离子水中并浸没总共4小时,在此期间小心地去除氟化盖玻片。聚合的聚丙烯酰胺表面用氩气干燥。
[1548]
随后将聚丙烯酰胺表面暴露于磷酸钠缓冲液(10mm,ph 8)中的硫醇化聚乙二醇(1kda)荧光素(fitc-peg-sh)和作为负对照的羧化聚乙二醇(1kda)荧光素(fitc-peg-cooh)1小时,然后依次用磷酸钠缓冲液(10mm,ph7)和含有0.05%tween20/0.5m nacl的相同缓冲液洗涤,以除去非特异性吸附的硫醇化和羧化荧光团。随后通过chemidoc(bio-rad)在荧光素泳道中对表面成像。
[1549]
结果和结论
[1550]
图38显示荧光信号,图39显示从聚丙烯酰胺凝胶表面测量的荧光,其中合并不同量的brapa,其暴露于fitc-peg-sh和fitc-peg-cooh。荧光素的固定仅在掺有brapa并且仅有硫醇化荧光素的聚丙烯酰胺表面成功,对羧化荧光素的非特异性吸附接近于零。
[1551]
与不含brapa(brapa 0%)的那些聚丙烯酰胺表面和含有brapa和羧化分子(fitc-peg-cooh)的那些聚丙烯酰胺表面相比,含有(brapa 0.1、0.2和0.3%)和仅来自巯基化分子(fitc-peg-sh)的聚丙烯酰胺表面获得了显著高的正荧光信号。结果表明,来自表面的溴乙酰基部分和来自荧光素标记分子的硫醇部分之间发生了特异性共价偶联。
[1552]
结果表明,分子,例如包括用于本发明方法的支持链和合成链的分子,可以容易地固定在与本文所述的多核苷酸合成反应相容的表面基底上。
[1553]
实例11.发夹dna寡聚体的表面固定和随后合并荧光标记的脱氧核苷三磷酸。
[1554]
本实例描述:
[1555]
(1)在薄的聚丙烯酰胺表面上呈现溴乙酰基的方法;
[1556]
(2)随后通过硫代磷酸酯官能化的发夹dna在有或没有接头下的共价偶联来固定发夹dna;和
[1557]
(3)将2'-脱氧核苷酸三磷酸(dntp)合并发夹dna中。
[1558]
所述方法几乎与任何类型的材料表面(例如金属、聚合物等)相容。
[1559]
(1):溴乙酰基官能化的薄聚丙烯酰胺表面的制备
[1560]
材料和方法
[1561]
首先在纯净的decon 90(30分钟)、水(30分钟)、1m naoh(15分钟)、水(30分钟)、0.1m hcl(15分钟)、水(30分钟)中通过超声处理清洁玻璃显微镜载玻片,最后用氩干燥。
[1562]
首先通过将1g丙烯酰胺单体溶解在50ml水中制备2%(w/v)丙烯酰胺单体溶液。将丙烯酰胺单体溶液涡旋并在氩气中脱气15分钟。将n-(5-溴乙酰胺基戊基)丙烯酰胺(brapa,82.5mg)溶解在825μl的dmf中,并加入到丙烯酰胺单体溶液中并进一步涡旋。最后,将1ml 5%(w/v)过硫酸钾(kps)和115μl纯四甲基乙二胺(temed)加入到丙烯酰胺溶液中,涡旋并将干净的玻璃显微镜载玻片暴露于所述丙烯酰胺聚合混合物中90分钟。90分钟后,用去离子水洗涤表面并用氩气干燥。在这个实例的下文中,这些表面将被称为“brapa改性表面”。作为负对照,通过排除向丙烯酰胺单体溶液中添加brapa溶液,也以与上述类似的方式制备不含brapa的聚丙烯酰胺表面。在这个实例的下文中,这些表面将被称为“brapa对照表面”。
[1563]
(2):硫代磷酸酯官能化的发夹dna在聚丙烯酰胺表面上的共价偶联
[1564]
材料和方法
[1565]
将具有4mm直径圆形开口的橡胶垫圈放置并固定在brapa改性表面和brapa对照表面上。首先用磷酸钠缓冲液(10mm,ph 7)引发表面10分钟。随后去除缓冲液,并将表面暴露于5'-荧光标记的(alexa 647)发夹dna寡聚体,分别具有和不具有以1μm浓度用六种和单一硫代磷酸酯修饰的接头,并在黑暗中培育1小时。brapa改性表面也与具有和不具有接头但没有硫代磷酸酯作为对照的dna寡聚体一起培育(在下文中称为“寡聚体对照表面”)。培育后,将表面用磷酸钠(100mm,ph7)冲洗,然后用tris-edta缓冲液(10mm tris,10mm edta,ph8)冲洗,最后用水冲洗。为了去除任何非特异性吸附的dna寡聚体,随后用含有1m氯化钠和0.05%(v/v)tween20的水洗涤表面,用水洗涤并用氩气干燥。在chemidoc成像仪上在alexa 647泳道中扫描表面。
[1566]
图40a显示了固定在不同样品上的没有接头的发夹dna序列。图40b显示了固定在不同样品上的具有接头的发夹dna序列。
[1567]
结果
[1568]
结果显示在图41和42中。图41显示源自发夹dna寡聚体的荧光信号,其中有和没有接头固定在溴乙酰基官能化的聚丙烯酰胺表面上,但不是来自brapa或寡聚物对照。
[1569]
图42显示了在聚丙烯酰胺表面上dna固定后测量的荧光强度。所述图显示了从各
种聚丙烯酰胺表面获得的表面荧光信号,并显示由于dna在溴乙酰基官能化的聚丙烯酰胺表面上成功的共价固定,与brapa和寡聚物对照表面(如(2)中所述)相比,从固定在brapa改性表面上的发夹dna寡聚体获得显著更高的信号。
[1570]
结论
[1571]
来自dna的荧光信号仅悬垂地存在于掺有brapa的brapa改性表面,表明通过硫代磷酸酯官能团将dna成功共价偶联到表面上。与没有接头的dna相比,从有接头的dna获得同源和更高的信号。
[1572]
(3):将三磷酸酯合并具有接头的发夹dna寡聚体中
[1573]
材料和方法
[1574]
将具有9mm直径的圆形开口的橡胶密封垫放在固定有具有接头的dna寡聚体的brapa改性表面上,并用合并缓冲液(50mm tris ph 8,1mm edta,6mm mgso4,0.05%tween20和2mm mncl2)引发10分钟。随后将表面暴露于含有dna聚合酶(0.5u/μl终止子x dna聚合酶)和三磷酸盐(20μm alexa488标记的dutp)的合并缓冲液中并培育1小时(此后在该实施例中称为“聚合酶表面”)。另外一组表面也暴露于没有终止子x dna聚合酶的合并缓冲液1小时作为负对照(在下文中称为“负表面”)。1小时后,将两种类型的样品在水中洗涤,随后暴露于含有1m氯化钠和0.05%(v/v)tween20的水中,并再次用水洗涤。在alexa 647和alexa 488泳道中使用chemidoc测量来自表面的荧光信号,以监测发夹dna(alexa 647)的存在和dutp(alexa 488)的合并。
[1575]
结果
[1576]
图43显示在合并alexa 488标记的dutp之前和之后从alexa 647和alexa 488泳道检测到的荧光信号。在合并之前和之后来自alexa 647的未改变的正信号表明表面固定的发夹dna在合并反应期间是稳定的,而来自alexa 488的正信号仅在合并反应后从聚合酶表面观察到,显示仅在聚合酶存在时成功合并dutp。
[1577]
图44显示了如(3)中所述在合并alexa 488标记的dutp之前和之后从
‘
聚合酶表面'和
‘
负表面'获得的alexa 647(发夹dna)和alexa 488(dutp)泳道中的测量荧光信号。由于成功合并,在聚合酶表面的合并反应后获得alexa 488荧光信号的显著增加,而由于不存在聚合酶,来自负表面的信号在合并反应后保持相同。在合并反应后,alexa 647泳道中的荧光信号基本保持不变,表明表面上存在发夹dna。荧光信号的轻微降低可能归因于第二轮曝光引起的光漂白效应。
[1578]
结论
[1579]
结果表明,包括用于本发明方法的支持链和合成链的分子,可以容易地固定在与本文所述的多核苷酸合成反应相容的表面基底上。结果进一步证明,这种分子可以接受新dntp的合并以延伸合成链,同时分子保持稳定并附着于基底。
[1580]
实例12.通过接头和硫代磷酸酯共价键连接固定于衍生化表面的发夹dna寡聚体的裂解和连接。
[1581]
该实施例描述了用接头与硫代磷酸酯官能化的发夹dna的衍生化表面的共价偶联,然后是裂解和连接反应。如实例11所述进行基底制备和发夹dna的偶联。
[1582]
(1):裂解具有接头的固定的发夹dna寡聚体
[1583]
材料和方法
[1584]
如实例11中所述,将发夹dna固定在表面brapa改性表面上。制备四组一式三份表面,包括用于裂解和连接反应的所有实验对照。实验条件如图45a所示。图45b显示固定在不同样品上的发夹dna的序列。
[1585]
在dna固定步骤后,将具有9mm直径圆形开口的橡胶垫圈放置在所有表面上,所述表面用在5'末端用alexa 647标记的dna固定,并用1x nebuffer 4(50mm醋酸钾,20mm tris-乙酸盐,10mm乙酸镁,1mm dtt,ph7.9)引发10分钟。注意,对于样品d,固定的发夹dna不含肌苷,肌苷被鸟嘌呤取代。随后将所有样品暴露于含有1.5u/μl核酸内切酶v的nebuffer 4(样品a、b和d)或不含核酸内切酶v的nebuffer 4(样品c)1小时。随后用1x t3 dna连接酶缓冲液(66mm tris-hcl,10mm mgcl2,1mm atp,7.5%peg6000,1mm dtt,ph 7.6)、含有1m氯化钠和0.05%(v/v)tween20的1x t3 dna连接酶缓冲液洗涤所有样品,再用1x t3 dna连接酶缓冲液洗涤,并在alexa 647泳道中在chemidoc成像仪上扫描。
[1586]
结果
[1587]
图46显示了在裂解反应之前和之后来自发夹dna寡聚体的荧光信号。
[1588]
图47显示了如上所述从dna固定表面获得的在裂解反应之前和之后测量的荧光信号。仅从样品a和b观察到成功的裂解反应,而由于序列中没有核酸内切酶v(样品c)或肌苷(样品d),样品c和d的荧光信号强度保持几乎相同。
[1589]
从样品a和b中观察到荧光信号的显著降低是由于在核酸内切酶v存在下在dna链内的肌苷位点的成功裂解反应。对于样品c和d,在dna内没有核酸内切酶v和缺乏肌苷分别引起荧光信号保持与dna固定后获得的初始信号几乎相同的水平。
[1590]
(2):连接反应
[1591]
材料和方法
[1592]
在如(1)中所述的裂解反应后,将样品a和b(如图45a中所述)暴露于含有mncl2(2mm)、在5'末端用alexa 647标记的肌苷链(16μm)和互补
‘
辅助'链(16μm)(序列显示在下面的图48中)的1x t3 dna连接酶缓冲液,用于样品a用t3 dna连接酶(250u/μl),对于样品b,没有t3 dna连接酶,作为负对照。将样品在各自的溶液中培育1小时。1小时后,将表面在水中洗涤,随后暴露于含有1m氯化钠和0.05%(v/v)tween20的水中,并再次用水洗涤。在alexa 647泳道中使用chemidoc测量来自表面的荧光信号。图48显示了用于连接反应的含肌苷链和互补“辅助”链的序列。
[1593]
结果
[1594]
图49显示了与连接反应监测有关的结果。在连接反应之前和之后从alexa 647泳道检测到荧光信号。连接后alexa 647泳道中荧光信号的增加仅从具有t3 dna连接酶的样品a获得,而由于不存在t3 dna连接酶,在样品b的连接反应后荧光信号保持在相同水平。
[1595]
图50显示,由于成功连接,样品a的连接反应后alexa 647荧光信号显著增加,其中信号水平恢复到如图47所示的dna固定后和裂解反应前的初始信号水平。由于不存在t3 dna连接酶,来自样品b的荧光信号在连接反应后保持相同。
[1596]
结论
[1597]
该实施例中的结果表明,包含用于本发明方法的支持链和合成链的分子,可以容易地固定在与本文所述的多核苷酸合成反应相容的表面基底上,并且可以在保持稳定以及附接于基底的同时进行裂解和连接反应。
[1598]
实例13.将3'-o-叠氮甲基-dntps合并平端dna的3'末端。
[1599]
该实例描述了将3'-o-叠氮甲基-dntps合并平端双链dna的3'末端。
[1600]
下面的步骤证明了通过dna聚合酶的酶促合并,将3'-o-保护的单核苷酸受控地添加到平端双链寡核苷酸中。这些步骤与图1至图5所示的合并步骤3一致。
[1601]
材料和方法
[1602]
材料
[1603]
1.内部合成的3'-o-叠氮甲基-dntps。
[1604]
2.新英格兰生物实验室设计的终止子x dna聚合酶具有增强的结合3-o-修饰dntp的能力。
[1605]
3.平端双链dna寡核苷酸。
[1606]
测试四种类型的可逆终止子:
[1607][1608]
方法
[1609]
1.将5μl的10x缓冲液(20mm tris-hcl,10mm(nh4)2so4,10mm kcl,2mm mgso4,0.1%x-100,ph 8.8,new england biolabs)与33.5μl无菌去离子水(elga veolia)在1.5ml eppendorf管中混合。
[1610]
2.将2μl的20μm引物(40pmol,1当量)(seq id:no:68,图51a)和3μl的20μm模板(60pmol,1.5当量)(seq id:no:69,图51a)添加到反应混合物中。
[1611]
3.加入3
’-
o-修饰的-dttp(2μl的100μm)和mncl2(2.5μl的40mm)。
[1612]
4.然后加入2μl的终止子x dna聚合酶(20u,new england biolabs)。
[1613]
5.将反应物在37℃培育30分钟。
[1614]
6.将反应物通过加入tbe-尿素样品缓冲液(novex)停止。
[1615]
7.将反应物在聚丙烯酰胺凝胶(15%)上用tbe缓冲液分离,并通过chemidoc mp成像系统(biorad)进行可视化。
[1616]
结果
[1617]
图51b描绘了一种凝胶,显示了在37℃存在mn2+离子的情况下,通过终止子x dna聚合酶合并3'-o-修饰的-dntp的结果。数据显示,终止子x dna聚合酶能够成功地将3'-o-修饰的dntps合并平端dna寡核苷酸的3'末端,从而产生一个碱基突出的碱基。
[1618]
实例14.支架多核苷酸的示例性裂解及其与多核苷酸连接分子的连接。
[1619]
该实例描述了在由通用核苷酸限定的裂解位点上对支架多核苷酸的裂解以及使用dna连接酶将多核苷酸连接分子与所裂解的支架多核苷酸的连接。该实例涉及具有平端的分子的连接,这与图1所示的发明版本1的合成方法一致。
[1620]
图52提供了描述dna合成反应循环的方案。该方案旨在与图1所示的发明版本1的合成方法一致。因此,图52中的方案显示了提供具有钝端的支架多核苷酸(方案最右边中间面板中的发夹结构),左链对应于支持链,右链对应于合成链。支持链的末端核苷酸与合成链的末端核苷酸配对。支持链的末端核苷酸在5'端包含磷酸基团。在该循环的下一步骤中,提供了多核苷酸连接分子(方案最下面部分的最右边的结构)。多核苷酸连接分子具有支持链(左链)和辅助链(右链)。多核苷酸连接分子具有互补的连接端,该末端具有平端并且包含2-脱氧尿苷(u)的通用核苷酸。辅助链在互补连接端的末端包含不可连接的核苷酸,并与支持链的末端核苷酸配对。支持链在互补连接端的末端核苷酸仅包含预定序列的核苷酸,描绘为“a”,仅用于说明。在将多核苷酸连接分子连接至裂解的支架多核苷酸后,在多核苷酸连接分子的互补连接端的支持链的末端核苷酸与支架多核苷酸的支持链的末端核苷酸连接,并且单链断裂(“缺口”)在多核苷酸连接分子的辅助链与支架多核苷酸的合成链之间产生。多核苷酸连接分子的支持链的末端核苷酸包含预定序列的核苷酸,并位于位置n。通用核苷酸因此占据位置n+1。在下一步骤中,在此情况下,仅出于说明目的,通过聚合酶或核苷酸转移酶的作用将另外的核苷酸(仅描述为“t”)并入支架多核苷酸的合成链中。所述另外的核苷酸包含可逆终止基团或封闭基团。另外的核苷酸与支持链中的配对核苷酸配对,在这种情况下是在连接步骤中合并的“t”核苷酸,从而形成核苷酸对。然后,该方案描述了脱保护或解封闭步骤,其中除去了可逆终止基或保护基。在图52所示的反应方案中,辅助步骤显示为在合并步骤之前作为可选步骤被去除。合并后,通过在位置n和n+1之间裂解支持链来裂解多核苷酸连接分子。裂解后,待合并的预定序列的第一和第二核苷酸,分别为“a”和“t”,以钝端排列形式保留在支架多核苷酸中,作为配对的核苷酸。
[1621]
如下所述,该实施例14描述了裂解支架多核苷酸并将多核苷酸连接分子与裂解的支架多核苷酸连接的步骤,如图52中的虚线所示。
[1622]
裂解步骤的材料和方法
[1623]
材料:
[1624]
1.在本实施例中使用的寡核苷酸是在室内设计的,并由integrated dna技术合成。这些在图53中进行了描述。
[1625]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1626]
方法:
[1627]
1.将39μl无菌蒸馏水(elga veolia)加入1.5ml eppendorf管中。
[1628]
2.然后将5μl 10x cut smart缓冲液(50mm醋酸钾,20mm tris-醋酸盐,10mm醋酸镁,100μg/ml牛,ph 7.9)加入同一eppendorf管中。
[1629]
3.将包含2'-脱氧尿苷(seq id:no 70)(20μmol/l)的1μl tamra(可以使用任何荧
光标签)发夹多核苷酸添加到同一试管中。
[1630]
4.然后将5μl user酶(尿嘧啶dna糖基化酶(udg)和核酸内切酶viii(neb)的混合物(1单/μl)添加到同一试管中。
[1631]
5.然后通过用移液管重悬将反应混合物轻轻地混合,以13,000rpm离心5秒,并在37℃培育30分钟。
[1632]
6.培育时间过去后,通过酶热灭活(即95℃10分钟)终止反应。
[1633]
7.培育时间过去后,使用qiagen核苷酸去除试剂盒纯化反应混合物。
[1634]
连接步骤的材料和方法
[1635]
材料:
[1636]
1.在本实施例中使用的寡核苷酸是在室内设计的,并由integrated dna技术合成。这些在图53中进行了描述。
[1637]
2.使用无菌蒸馏水(elga veolia)将寡核苷酸稀释到100μm的储备浓度。
[1638]
方法:
[1639]
使用以下步骤进行寡核苷酸的连接反应:
[1640]
1.将来自上述裂解反应的13μltamra(可使用任何荧光标签)裂解的平端多核苷酸添加到1.5ml eppendorf管中。
[1641]
2.然后将30μl 2x t3 dna连接酶反应缓冲液neb(132mm tris-hcl,20mm mgcl2、2mm二硫苏糖醇,2mm atp,15%聚乙二醇(peg6000)并且ph为7.6,25℃)和2μl 40mm mncl2添加到同一eppendorf管中。
[1642]
3.将5μl 2-脱氧尿苷(u)链(200μmol/l)(seq id:no 71)和5μl辅助链(200μmol/l)(seq id:no 72)添加到同一试管中。
[1643]
4.将5μl t3 dna连接酶neb(3000单位/μl)加入同一试管中。
[1644]
5.然后将反应混合物在室温下培育30分钟。
[1645]
6.在培育时间过去之后,通过添加tbe-尿素样品缓冲液(novex)终止反应。
[1646]
结果
[1647]
结果示出在图54中。
[1648]
在上述实施例中,seq id no 1-72中所示的所有寡核苷酸在3'末端具有羟基。除了seq id no 7、seq id no 18、seq id no 35和seq id no 70之外,seq id no 1-72中所示的所有寡核苷酸在5'末端缺少磷酸基团。
[1649]
应当理解,所公开方法和产物的不同应用可以根据本领域的特定需要而定制。还应理解,本文所使用的术语仅出于对本发明的特定实施例进行描述的目的而并不旨在是限制性的。
[1650]
如在本说明书和所附权利要求中所使用的,单数形式“一(a)”、“一(an)”和“所述(the)”包含复数指示物,除非上下文另外明确指明。因此,例如,提及“多核苷酸连接分子”包括两个或更多个这样的多核苷酸,提及“支架多核苷酸”包括两个或更多个这样的支架多核苷酸,等等。
[1651]
本文所引用的所有公开、专利和专利申请特此通过引用整体合并。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1