用于合成单金属纳米桥结构的方法以及通过使用其制造DNA点突变检测传感器的方法
用于合成单金属纳米桥结构的方法以及通过使用其制造dna点突变检测传感器的方法
技术领域
1.本发明涉及一种包含金属纳米桥结构的单纳米粒子生物传感器平台及一种构建单纳米粒子生物传感器平台方法。更确切地说,本发明涉及一种包含金属纳米粒子的单纳米粒子生物传感器平台,每一个金属纳米粒子由两个金属纳米晶种(nanoseed)及固定于所述金属纳米晶种之间的生物分子组成;包含单纳米粒子生物传感器平台的生物传感器;使用生物传感器检测突变的方法;以及用于构建单纳米粒子生物传感器平台的方法,包含形成金属纳米粒子,每一个金属纳米粒子由两个金属纳米晶种及固定于所述金属纳米晶种之间的生物分子组成。
背景技术:
2.许多疾病具有遗传成分;因此其检测需要清晰的了解生物医学诊断学中由突变引起的代谢病症。诊断基因突变的大部分方法依赖于测序,但采集突变信息的最佳方法可在没有序列的先验知识,理想地在无标记和活体外环境的假影的情况下判定突变碱基的存在和一致性。已努力找到一种具有与许多生物体中复制后错配修复(mmr)相反的系统的方法,其中根据利用纳米等离子(nanoplasmonic)生物传感器引入修复mutl和突变dna的其它酶,错配修复(mismatch repair,mmr)起始蛋白质muts以序列非特异性方式识别突变(马,x.(ma,x.),特龙,pl(truong,pl),安,n.h.(anh,n.h.)以及希姆,s.j.(sim,s.j.),《生物传感器与生物电子学(biosensors&bioelectronics)》67,59
‑
65(2015))。
3.纳米生物感测的可靠性通常由两个主要因素来确定:用于单一生产的纳米材料和用于靶标识别的生物分子,其与特定物理条件的检测灵敏度及选择性直接相关。在这些纳米材料当中,等离子体纳米粒子由于其与入射光相互作用并产生局部化表面等离子共振(lspr)的能力而引起人们的兴趣。纳米结构中的电子以给定共振频率的集体振荡将局部折射率(ri)的变化转换为其吸收光谱和散射光谱的等离子带的偏移。感测尺寸可减小至单纳米粒子,进而有助于单纳米粒子感测(snps);这类单np感测(single
‑
nanoparticle sensing,snps)可以纳米尺寸传递局部生物信息,其中检测极限(lod)使用极小的感测体积达到可数数量的分子。相比之下,使用大量溶液或平坦表面的大部分其它感测技术展现出定位且分离感测元件的有限能力,并且受到缓慢的分子扩散、随机结合以及络合生物分子的频繁解离及随之而来的反应不平衡的限制,从而导致信号以低信噪比(signal
‑
to
‑
noise,s/n)波动。snps传感器为能够实现高通量及并行读数的微型探针,其中np的结构和局部化感测体积/面积对于ri灵敏度来说是必需的。对具有不同形状的金纳米粒子(np)的ri灵敏度的系统性研究表明棒状np展现出对ri变化的最高灵敏度(特龙,p.l.,马,x.以及希姆,s.j.,《纳米尺寸(nanoscale)》 6,2307
‑
2315(2014))。近年来,已证实除了粒子形状之外,纳米间隙和纳米桥结构相关联并且通过等离子耦合产生较强的光信号,进一步增强局部场以产生不同的光谱响应(利姆,d.k. (lim,d.k.)等人.《自然纳米技术(nature nanotech.)》6,452
‑
460(2011);纳姆j.m.(nam,j.m.),欧,j.w.(oh,j.w.),李,h.(lee,h.)
以及苏荷,y.d.(suh,y.d.),《化学研究报告(acc.chem.res.)》49,2746
‑
2755(2016))。然而,合成具有预定结构的胶状等离子np 具有挑战性,这是因为难以处理溶液中的瞬态原子。此外,化学合成的np被限制为具有相同表面特征的高度对称形状(例如,纳米球、纳米棒、纳米立方体、纳米盘以及其它)。np的结构可编程性可为克服snps在灵敏度和可再生性方面的局限性提供强有力的手段。近年来,两个研究小组在使用可编程生物分子(即,dna)以低于5纳米精密度进行设计合成方面获得突破,进而通过在dna模具中铸造(孙,w.(sun,w.,)等人.《科学(science)》346,1258361 (2014))或使用dna框架(马,x.,等人.《自然通讯(nat commun)》7,(2016))产生精细定义的纳米等离子体粒子。
4.snps提供多种利用其与例如核酸或蛋白质的生物分子尺寸相当的大部分固有较小感测体积的应用。举例来说,muts蛋白质为因此,muts吸附于单np上可大幅度改变其表面电子的集体振荡行为,从而导致np光谱中的波长偏移。因为单点突变实质上不基于pcr和其它基于dna芯片的检定来识别,所以包含错配dna的muts的生物相互作用的特异性促进了关于核苷酸多态性的研究。最广泛的研究是基于单分子荧光共振能量传递法 (smfret);然而,这些需要劳动密集型步骤,例如标记muts或制造用于dna的放射性探针。通过原子力显微镜使用单分子成像的最新研究是极复杂的并且几乎不可能适用于生物医学诊断。在另一方面,通过凝胶迁移率变化分析、滤波器/芯片结合分析、核酸酶保护分析、表面等离子共振(spr)、电化学分析以及石英晶体微天平(qcm)对单点突变进行大量测量并不输出关于分子相互作用的实时信息并且为低效及费时的。
5.因此,本发明的发明人已较早地并且集中地实施研究以解决现有技术的问题。因此,本发明人通过修改各dna分子的两端以便结合至金纳米晶种并且以两个相反的方向使金结晶来成功的制备具有高ri灵敏度的au桥接纳米粒子;以及发现包含au桥接纳米粒子的单纳米粒子生物传感器平台可用于以高灵敏度及可靠性检测靶标并且根据muts针对突变的相对活性而直接鉴别出各种突变。已基于这个发现实现了本发明。
技术实现要素:
6.本发明要解决的问题
7.本发明的一个目标是提供一种包含金属纳米桥结构的单纳米粒子生物传感器平台和一种包含单纳米粒子生物传感器平台的生物传感器。
8.本发明的另一目标是提供一种使用生物传感器检测突变的方法。
9.本发明的另一目标是提供一种用于构建单纳米粒子生物传感器平台的方法,包含形成金属纳米粒子,每一个金属纳米粒子由两个金属纳米晶种及固定于所述金属纳米晶种之间的生物分子组成;以及使用所述金属纳米粒子形成金属纳米桥结构。
10.解决问题的方法
11.本发明提供一种包含金属纳米桥结构的单纳米粒子生物传感器平台和一种包含单纳米粒子生物传感器平台的生物传感器。
12.本发明还提供一种使用生物传感器检测突变的方法。
13.本发明还提供一种用于构建单纳米粒子生物传感器平台的方法,包含形成金属纳米粒子,每一个金属纳米粒子由两个金属纳米晶种及固定于所述金属纳米晶种之间的生物分子组成;以及使用所述金属纳米粒子形成金属纳米桥结构。
14.本发明的效果
15.本发明的单纳米粒子生物传感器平台可用于不仅以高灵敏度及可靠性检测靶标,而且直接鉴别各种突变,从而实现突变的有效诊断。因此,本发明的单纳米粒子生物传感器平台可用于广泛范围的领域,包含生物医学诊断。
附图说明
16.图1示出单np的ri灵敏度分析的示意性图示。lspr波长响应于周围介质的ri变化而偏移。右侧的表示出介质的ri值。
17.图2示出auns
‑
dsdna
‑
auns的制备和表征:a通过电泳分离的不同auns
‑
ssdna共轭物的凝胶图像。各条带表示固定到一个auns的一定数量的ssdna。最左边的条带表示未结合dna的裸纳米晶种;b ssdna杂交之后通过电泳分离及鉴别的auns
‑
dsdna
‑
auns的凝胶图像;以及c auns
‑
dsdna
‑
auns的tem图像。
18.图3示出snps系统:a基于通过白光照射进行单au桥接np的rls及lspr的snps 系统的详细配置;b检测腔室的示意图;c通过相机获取的腔室图像。标记粒子间间距比闪光点直径大约5倍的个别纳米粒子的位置并对所述个别纳米粒子进行分析;d腔室的sem图像;e每分钟一次持续10分钟获取的au桥接np的原始光谱;以及f 10个原始光谱的洛伦兹拟合(lorentzian fitting)显示出0.188纳米的峰值测量精密度。
19.图4示出感测特异性:a muts与探针的对照实验。单np的rls光谱显示无明显的λ
max
偏移(0.356纳米蓝移);b muts与homodna的对照实验显示λ
max
的0.343纳米红移;c突变靶dna(mdna)与血清中的非特异性组分的对照实验。rls光谱显示无明显的λ
max
偏移(0.700 纳米红移);d将muts引入至血清中在与c中相同的检测条件下显著产生14.7纳米红移。
20.图5示出显示出限制酶的活性位点的序列。mboi,5
′‑
gatc
‑3′
;alui,5
′‑
agct
‑3′
;styi, 5
′‑
ccwwgg
‑3′
。
21.图6示出在酶mboi、alui以及styi限制性消化之后的不同细胞系的brca1的计算分段图谱。使用软件genetyx 4.0(genetyx公司(genetyx corporation),日本东京(tokyo,japan)) 进行分析。
22.图7示出三种限制酶的协同性消化前后的brca1 dna的琼脂糖凝胶电泳的结果(显示于最右侧的dna阶梯)。从乳癌细胞系mcf7和hcc1937,以及卵巢癌细胞系snu251中萃取基因组dna。
23.图8示出个别纳米粒子的等离子兴趣场(foi):a foi及负载au桥接np的muts的可用空间的示意图;b ccd图像的比例校准。一个像素的长度等于0.42微米;c foi的高度(h) 的图示;以及d lspr峰偏移周期性的图示。以绿色标记的区域为短程折射率感测区,其中等式δλ
max
=mδn[1
‑
exp(
‑
2d/l)]可用于描述随介质浓度变化而变的lspr波长偏移。因为这个等式并不适用于长程lspr感测,所以示意性曲线并不考虑长程lspr中的振荡行为的实际特征概况(例如,不调和特性)。
[0024]
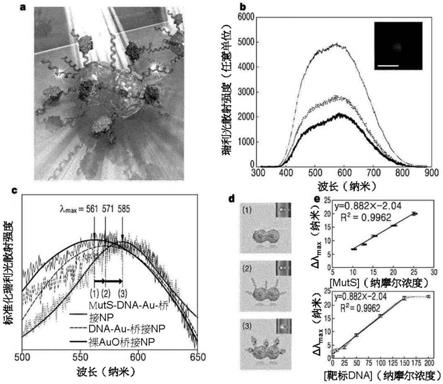
图9示出具有高灵敏度和保真度的单纳米粒子(np)生物传感器的制造:a用于鉴别单点dna突变的snps的示意性图示;b au桥接np的代表性瑞利光散射(rayleigh lightscattering;rls)光谱及原位暗场显微镜图像。比例尺,1微米;c在dna与muts结合后的局部化表面等离子共振(lspr)λ
max
偏移。(b,c)使用相同线图例;d分子结合的每个步骤 (1、2以
及3)。插图:用ccd相机获得的单np的实时图像;e用于适当信号响应的muts 浓度。校准曲线示出δλ
max
与各种muts浓度之间的线性关系;以及f突变dna靶标的lod。校准曲线示出δλ
max
与各种靶dna浓度之间的线性关系。
[0025]
图10示出每个au桥接np的探针平均负载量(n
*
)的估算。将纳米粒子建模为两个由圆柱体(黄色)桥接的球体(灰色);假定dna足迹以最高负载密度和彼此最近的距离均匀地分布于粒子上,且由此建模为球体上的圆和圆柱体上的椭圆。“有效负载边缘”线上方的表面积与总面积的比率近似于(180
°‑
73
°
)/180
°
=59.4%。
[0026]
图11示出等离子体纳米粒子(np)于溶液中的设计合成:a经设计np模型(上图;尺寸单位,纳米)和等离子共振电场模式(下图)的图示;b线性拟合局部化表面等离子共振 (lspr)波长偏移与np周围环境的ri变化;c示出通过用nh3oh
+
还原aucl4‑
进行au原子的方向特定、轮廓跟随以及形状控制式结晶的早期阶段的示意图。在ph 5及4的合成条件下, dna的水界面提供精确的可控性,从而产生分别具有纳米桥和纳米间隙的np,如tem图像中所示。比例尺,20纳米;d auns及au桥接np的x射线衍射光谱;以及e dna定向纳米晶体的hr
‑
tem图像及选定区域的快速傅里叶变换模式(右侧)。比例尺,10纳米。
[0027]
图12示出au桥接np的尺寸分布。使用imagej测量纳米结构的长度及直径。统计结果显示狭窄尺寸的分布。
[0028]
图13示出八个单点突变的可鉴别检测:a对于muts结合于各突变dna及同源双链体的实时监测;以及b muts与各点突变之间相互作用的速率常数的重绘曲线。
[0029]
图14示出感测的可靠性:a对muts以各种浓度结合于gt
‑
突变dna的实时监测;以及 b速率常数对muts浓度的依赖性。
[0030]
图15为针对不同点突变的muts亲和力的图谱。通过单au桥接np感测muts与八个不同突变之间可数数量的结合事件来创建图谱。将来自人类乳癌细胞hcc1937及卵巢癌细胞 snu251的brca1中关于潜在点突变的存在及类型的信息的诊断结果输入到图谱中,从而分别预测+c及a
‑
c的突变。
[0031]
图16示出用本发明中研发的八个snps芯片对brca1点突变的诊断结果:a
‑
h将来自人类乳癌细胞系hcc1937的样本作为分析物进行检测。来自mcf7的样本用作对照。临床样本在监测期间产生较大的δλ
max
值。除了5382insc以外,无芯片产生有效k
reaction
,表明分析物含有单个胞嘧啶复制。
[0032]
图17示出使用5382insc探针对来自细胞系hcc1937的brca1中的突变进行检测。全部实验是一式三份地进行。将k
reaction
平均值(0.0573)输入到针对不同点突变的muts亲和力图谱中,从而预测靶标中+c的突变。
[0033]
图18示出dna测序结果。与细胞系mcf7的brca1序列(没有点突变的阴性对照)相比,hcc1937序列展现出单个胞嘧啶插入。
[0034]
图19示出来自卵巢癌细胞系snu251的dna样本的用户指定基因组区域中点突变的诊断。分析位于染色体17的长臂的区域2条带1上的43047665处的靶标,一式三份。将k
reaction
平均值(0.0320)输入到针对不同点突变的muts亲和力图谱中,从而预测靶标中ac的突变。插图示出对来自mcf7细胞的样本进行检测的对照实验的结果。
[0035]
图20示出dna测序结果。与细胞系mcf7的brca1序列(没有点突变的阴性对照) 相比,snu251序列展现出单个g>a取代。
具体实施方式
[0036]
除非另外定义,否则本文中所使用的所有技术和科学术语具有与本发明所属领域的技术人员通常理解的含义相同的含义。通常,本文中使用的术语为熟知的且通常在所属领域中采用。
[0037]
在一个方面,本发明涉及一种金属纳米桥结构,具体地包含金属纳米粒子的金属纳米桥结构,每一个金属纳米粒子由两个金属纳米晶种(nanoseed)及固定于所述金属纳米晶种之间的生物分子组成;以及一种包含金属纳米桥结构的单纳米粒子生物传感器平台。
[0038]
在另一方面,本发明涉及一种用于构建单纳米粒子生物传感器平台的方法,包含形成金属纳米粒子,每一个金属纳米粒子由两个金属纳米晶种及固定于所述金属纳米晶种之间的生物分子组成;以及使用所述金属纳米粒子形成金属纳米桥结构。
[0039]
在本发明中,金属优选地是由以下组成的群组中选出:金(au)、铜(cu)、铂(pt)以及钯(pd),优选地为金(au)。
[0040]
在本发明中,金属纳米晶种是由以下组成的群组中选出:纳米球(nanosphere)、纳米棒 (nanorod)、纳米棱柱(nanoprism)以及纳米板(nanoplate)。
[0041]
在本发明中,生物分子是由以下组成的群组中选出:单链dna、双链dna、dna寡聚物、rna寡聚物、质粒dna、多肽以及蛋白质,优选地为双链dna。
[0042]
在本发明中,金属纳米晶种优选地具有25纳米或小于25纳米的直径。
[0043]
在本发明中,生物分子优选地具有30纳米或小于30纳米的长度。
[0044]
本发明方法更包含在金属纳米粒子的表面上用还原剂还原金属离子以生长金属纳米粒子。
[0045]
在本发明中,还原剂为羟胺(nh2oh),但不限于此。
[0046]
在另一方面,本发明涉及一种包含单纳米粒子生物传感器平台的生物传感器。
[0047]
本发明的生物传感器包含蛋白质,优选地错配修复起始蛋白质(mismatch repair protein, muts)。muts是指识别核酸分子中的错配并且可结合至错配位点的蛋白质。muts另外还包含野生型蛋白质,所述野生型蛋白质具有其中取代、缺失、添加和/或插入一或多个胺基酸的胺基酸序列,只要其可识别错配即可。
[0048]
本发明的生物传感器具有比纳米棒高的折射率(refractive index,ri)灵敏度。ri灵敏度定义为lspr峰偏移相对于粒子周围介质的折射率变化的相对变化。在下文的实例部分中,根据本发明的金属纳米桥结构的ri灵敏度经确认高于纳米棒的ri灵敏度,已知所述纳米棒的 ri灵敏度高于其它纳米结构的ri灵敏度。
[0049]
本发明的生物传感器用以检测突变,尤其点突变。
[0050]
本发明的生物传感器用以指定样本中brca1突变的类型。
[0051]
在又一方面,本发明涉及一种使用生物传感器检测突变,尤其点突变的方法。
[0052]
本发明方法用以通过错配修复起始蛋白质(muts)与突变核酸分子的结合分析来鉴别蛋白质,优选地突变。
[0053]
用于实施本发明的模式
[0054]
[实例]
[0055]
将参考以下实例更具体地解释本发明。本领域的技术人员应了解,这些实例仅为示出性的且本发明的范围不理解为限于所述实例。因此,应通过所附权利要求和其等效物
来限定本发明的真实范围。
[0056]
实例1:构建单纳米粒子感测平台及使用单纳米粒子感测平台检测点突变
[0057]1‑
1:材料
[0058]
使用金纳米晶种(auns;5纳米)溶液(英国生物细胞国际公司(british biocell international),英国克拉姆林(crumlin,uk))、洗涤/存储缓冲液(具有0.02%nan3、0.01%tween 20、0.1%bsa的10毫摩尔浓度pbs,ph 7.4;产品目录#:wb
‑
100,美国大洋纳米科技公司 (ocean nano tech),美国加利福尼亚州圣地亚哥(san diego,ca,usa))、二硫苏糖醇(dtt,普洛麦格公司(promega),美国威斯康星州麦迪逊(madison,wi,usa))以及限制酶styi (#r648a,普洛麦格公司,美国威斯康星州麦迪逊)、离心机(及颇尔生命科学有限公司(pall life sciences,inc),美国密歇根州安娜堡(ann arbor,mi,usa))以及 2
‑
{2
‑
[2
‑
(2
‑
{2
‑
[2
‑
(1
‑
巯基十一
‑
11
‑
基氧基)
‑
乙氧基]
‑
乙氧基}
‑
乙氧基)
‑
乙氧基]
‑
乙氧基}
‑
乙胺盐酸盐(oeg;科斯生物技术(cos biotech),韩国大田(daeieon,korea))。衍生自嗜热性细菌栖热水生菌(thermus aquaticus)的muts蛋白质是由日本基因公司(nippongene co.)(日本东京)供应并且存储在
‑
20℃下的含有100毫摩尔浓度nacl、0.1毫摩尔浓度edta、1毫摩尔浓度dtt以及50%丙三醇的20毫摩尔浓度tris
‑
hcl缓冲液(ph 7.5)中。g
‑
spin
tm
总dna萃取试剂盒(#17046)是由intron生物技术公司(intron biotechnology)(韩国京畿道(gyeonggi, korea))供应。限制酶mboi(#r0147)及alui(#r0137)是获自新英格兰生物实验室(neb) (英国赫特福德郡希钦(hitchin,hertfordshire,uk))。肝糖(#901393)是获自罗氏公司(roche) (美国印第安纳州印第安纳波利斯(indianapolis,in,usa))。聚(乙二醇)甲基醚硫醇(peg, mn=800)、杂交缓冲液、含有单股结合蛋白的杂交洗涤包以及全部其它化学反应剂购自西格玛
‑
奥德里奇公司(sigma
‑
aldrich)(美国密苏里州圣路易斯(st.louis,mo,usa))且不经进一步纯化即使用。实验中使用的全部玻璃器皿在王水溶液(aqua regia solution)中清洗并且在使用之前用超纯水(18.2mω
·
cm
‑1)充分地冲洗。使用的全部寡核苷酸来自整合dna技术公司(integrated dnatechnologies)(美国衣阿华州科勒尔维尔(coralville,ia,usa))。含有点突变的8个dna靶标的序列描述于表1中。相应的同源双链体(完全匹配)序列如下: attgaaagttgcagaatctgcccagagtccagctgctgctcatactactga。单链dna (ssdna)的指定名称及信息显示于表2中。dna靶标及探针的序列显示于表2中。
[0059][0060]
[表2]
[0061][0062]1‑
2:np建模及数字模拟
[0063]
执行具有球形、棒状以及二聚体形状的纳米结构的建模及光学模拟并且使用软件 comsol桥接np。np是由au组成;粒子尺寸(dimension)设定为均匀的以便于进行比较。根据实验中合成的产物测定最终尺寸。在局部介电环境中执行光学模拟,其中制备不同重量比的水
‑
甘油混合物以使得周围介质的ri介于1.333至1.443范围内(图1)。
[0064]1‑
3:auns与ssdna共轭
[0065]
将全部5
′
硫醇修饰的寡核苷酸用1∶100比率的寡核苷酸od比dtt溶液培育15分钟并且用乙酸乙酯纯化两次。5
′‑
硫醇的双硫键裂解成活性巯基形式并且立即与au表面共轭。在与溶液中的dna共轭之前,auns涂布有双(对磺酸根苯基)苯基膦二水合物二钾(bspp;100毫升auns溶液与100毫克bspp混合10小时)以提高auns对高度离子环境的耐受性。随后将auns溶液与nacl混合,这会使得颜色从暗红色变为淡紫色。溶液以500
×
克离心30分钟且将沉淀物再悬浮于1毫升的0.5毫摩尔浓度bspp中。在添加0.5毫升甲醇后,溶液的颜色再次从暗红色变为淡紫色;通过离心(30分钟,500
×
克)收集auns并将其溶解于1毫升0.5
×ꢀ
tbe缓冲液中。将auns的浓度增加到若干微摩尔浓度,如用紫外光
‑
可见光
‑
近红外光谱仪 (uv
‑
3600;日本京都岛津(shimadzu,kyoto,japan))所测量;根据制造商的说明书,1 od的 5纳摩尔浓度auns等于每微升有5.00
×
10
13
个粒子。在室温下,将auns与ssdna
‑
1以1∶1 的化学计量比在含有50毫摩尔浓度nacl的0.5
×
tbe缓冲液中培育整夜。之后,将60%甘油添加到溶液中以获得最终10%甘油混合物以防止auns
‑
ssdna在凝胶电泳期间在缓冲液中扩散。在10 v/cm下持续1小时,具有不同数量的结合ssdna的auns在0.5
×
tbe缓冲液中的 3%琼脂糖凝胶上分离成不同的条带(图2)。将对应于auns与ssdna的一条链共轭的条带 (auns
‑
1ssdna
‑
1)在0.5
×
tbe缓冲液中培育以供进一步使用。进行相同的培育及分离程序以使auns与ssdna
‑
2共轭,获得auns
‑
1ssdna
‑
2。
[0066]1‑
4:纳米结构的定向合成
[0067]
将金前体(haucl4,0.03%)及还原剂(nh2oh
·
hcl,1毫摩尔浓度)单独地溶解于水中并且在氮气环境下通过逐步地添加naoh将各溶液的ph调节至5或4(
±
0.1)。用于dna定向合成的晶种是通过auns
‑
1ssdna
‑
1与auns
‑
1ssdna
‑
2杂交呈auns
‑
dsdna
‑
auns形式来产生。为增加杂交效能,将两种共轭物于0.5
×
tbe中的相等体积混合并添加nacl以使离子强度增加100毫摩尔浓度。将混合物在37℃下振荡整夜并且通过凝胶电泳用如上文所描述的相同程序来分离auns
‑
dsdna
‑
auns(图2)。将含有auns
‑
dsdna
‑
auns的凝胶浸没于50毫升最终peg/晶种摩尔比为100∶1的洗涤/存储缓冲液中。通过离心管(分子量截止值(molecularweight cut
‑
off)30k,3000
×
克)纯化并且浓缩溶液,且因此使得晶种受到中性peg层保护
以提高稳定性并且减少带电分子于auns表面上的非特异性吸附。将晶种与金前体轻缓地搅拌 10分钟,最终浓度为2纳摩尔浓度;将10微升溶液与17.54微升还原剂混合,并且在1分钟内观察到颜色从无色变为淡红色。在15分钟之后,通过在水中重复再悬浮及离心来洗涤合成的np。在300千伏加速电压下以z
‑
对比度(contrast)及次级电子模式获得(hd2300;日立 (hitachi))np的tem及hr
‑
tem图像。制备样本以使用染色板(电子显微科学(electronmicroscopy sciences))及带碳膜的400
‑
目铜网(泰德佩拉公司(tedpella))进行tem。使用软件imagej测量tem平面中纳米结构的长度及直径。比例尺为20纳米及50纳米的图像示出足够大以进行精密测量的纳米结构。粒子产率计算为au桥接np与总粒子的比率。非所要粒子可容易地区分为超大或过小的桥接纳米粒子及由auns
‑
ssdna生长的纳米球,所述纳米球在合成反应期间是由auns
‑
dsdna
‑
auns变性(马,x.等人,《自然通讯(nat commun)》 7,12873,2016)。用数字化显微图软件(加坦公司(gatan),美国加利福尼亚州普莱森顿 (pleasanton,ca,usa))分析hr
‑
tem图像的快速傅里叶变换模式以确认结晶结构及生长取向。
[0068]1‑
5:snps平台设置
[0069]
snps系统的总体配置展示于图3a中。为构建检测腔室,将显微镜载玻片(22
×
40
×
0.1 毫米;华纳仪器(warner instruments))涂布有第一接触清洁聚合物(光子清洁技术(photoniccleaning technologies)),所述第一接触清洁聚合物在固化15分钟后立即剥离掉。将载玻片用王水溶液冲洗整夜;在用超纯水冲洗之后,将载玻片浸没于5%(v/v)3
‑
氨基丙基三乙氧基硅烷/纯乙醇中15分钟,接着在超纯水中超声处理5分钟。这个过程重复三次。将3微升体积的经稀释au桥接np溶液(od~0.05)以液滴形式添加于硅烷化载玻片上,接着在室温下培育 1分钟。随后在生物防护罩中用超纯水及乙醇洗涤载玻片以最小化气载残渣的污染,并最终用氮气吹干。将载玻片放置在封闭槽成像微流体腔室(rc
‑
30;华纳仪器)中,所述腔室组装于平台控制器(三月屋传感技术公司(marzhauser sensotech),德国韦茨拉尔(wetzlar,germany)) 上并且与实现快速扰动溶液混合的流动装置以及流速控制系统(phd 2000;哈佛设备(harvardapparatus),美国马萨诸塞州霍利斯顿(holliston,ma,usa))连接。用感测系统获得腔室中各np的图像及瑞利散射特性(图3b)。用彩色相机(d50;尼康(nikon),日本东京(tokyo, japan)获得配备有100w卤素来源(型号7724,菲利普斯(philips))、暗场干聚光器(na= 0.80
‑
0.95;尼康)以及100
×
物镜(cfi plan fluor elwd,na=0.6;尼康)的倒置显微镜(eclipsete2000
‑
u;尼康)视场的图像,仅分析粒子间间距比闪光点直径大约5倍的个别纳米粒子以最小化粒子间共振耦合的效应(图3c)。使腔室的图像在
‑
70℃聚焦于电荷耦合装置(ccd) 相机(pixis:400b;普林斯顿仪器(princeton instruments),美国新泽西州特伦顿(trenton,nj, usa))上,帧积分时间为100毫秒。将显微镜及长通滤波器的输出端口的光束分光器放置于所述ccd之前。平台允许在18℃下在暗室中使用rls光谱图(microspec 2300i;罗珀科学 (roper scientific),法国吕弗斯街南山8号(montagne sud
‑
8,rue du forez,france))测定腔室中各np的瑞利光散射(rls)特性。记录300
‑
900纳米范围内的光谱,采集时间为1秒。用洛伦兹演算法拟合光谱数据以消除噪音,并使用origin2018软件(originlab公司,美国马萨诸塞州北安普敦(northampton,ma,usa))测定精确的λ
max
。应用这一方法分析每分钟从相同纳米粒子获取一次的10个光谱,得到限制拟合峰测量精确值为0.188纳米(图3e及f)。峰位置的波动归因于包含以下的仪器
因素:光谱仪分辨率、腔室的物理均一性、温度的瞬时变化以及液体的流速;及例如以下的分析因素:显微镜聚焦控制、光谱源校正、曝光像素选择以及空间平均化。除0.188纳米的波动以外,随机检测168个纳米粒子中的整体实验峰不确定性为0.487纳米,其中尺寸、形状以及取向的纳米粒子因素导致0.299纳米差异。
[0070]1‑
6:点突变的检测
[0071]
将玻璃载片安装在snps平台中之后,通过注入75%乙醇来冲洗腔室5分钟,接着用洗涤 /存储缓冲液冲洗20分钟以移除污染物及未结合的au
‑
np。在拍摄腔室之后,记录au桥接 np的位置。一个np代表一个检测集且针对分子结合的每个步骤测定其光学特性。腔室在室温下装填有100纳摩尔浓度的探针dna(例如,探针
‑
gt)持续8小时且在杂交缓冲液中引入不同浓度的靶标dna(例如4956a>g)之前用洗涤/存储缓冲液冲洗5分钟。通过用杂交洗涤包进行冲洗来移除未结合的靶标,之后注入目标浓度的muts溶液。在18℃下在结合缓冲液(ph 7.5;100毫摩尔浓度nacl、1毫摩尔浓度dtt、0.1毫摩尔浓度edta以及5毫摩尔浓度mgcl2)中以1微升/分钟的流速进行muts与dna的结合。针对典型检测,在腔室中通过探针
‑
gt俘获100纳摩尔浓度的靶标dna并使其与20纳摩尔浓度的muts蛋白质反应2 分钟。使用winspec软件(罗珀科学(roper scientific))记录并处理ccd相机对单np的实时成像及rls光谱。实施相同检测条件下的对照实验以研究muts与探针(无靶标结合)及 dna同源双链体(同源dna;无突变)的相互作用。针对关于dna与非特异性蛋白质(无 muts)相互作用的研究,在注入具有gt突变的靶标dna之后,引入人类血清。光谱分析之后,在95℃下用洗涤/存储缓冲液冲洗腔室30分钟以移除蛋白质。在捕获相同靶标之后,将含有20纳摩尔浓度muts的相同血清溶液注入腔室中,且随后再次记录光谱以用于进一步分析(图4)。
[0072]1‑
7:制备及检测来自细胞系的样本
[0073]
使用g
‑
spin
tm
总dna萃取试剂盒萃取基因组dna并在纯化及进一步限制性消化之前在 55℃下用200纳克/毫升蛋白酶k及10纳克/毫升rnase a处理30分钟。用限制酶mboi、alui 以及styi进行消化以产生50
‑
60个碱基对核苷酸。详细地说,由mboi及alui进行消化得到100
‑
500个碱基对的片段。由于brca1中存在styi位点,所以再由styi将片段消化成长度为~ 50个碱基对的目标样本。酶的特异性位点及计算的分段图谱可见于图5和图6中。为有效收集dna,在乙醇沉淀期间添加肝糖,接着以13,000转/分钟离心15分钟。使用nano
‑
200微型光谱仪dc24v(#as
‑
11030
‑
00;奥盛仪器(allsheng instruments),中国杭州)评定dna 的浓度及纯度。通过凝胶电泳评估dna的完整性,其中在4℃下将300纳克dna样本以2.5 v
·
cm
‑
1装载于0.7%琼脂糖凝胶上,用0.5%溴化乙锭染色,以及通过uv照射用davinch
‑
geltm凝胶成像系统(扬华科技公司(young wha scientific),韩国首尔(seoul,korea))进行检测 (图7);在90℃下将30微升核苷酸解链成单链靶标中1分钟且在70℃下在5分钟内注入装填有杂交缓冲液的snps腔室中。随后,利用杂交洗涤包冲洗腔室,之后在18℃下以1微升/分钟的流速将20纳摩尔浓度的muts溶液引入结合缓冲液中。为诊断来自snu251细胞系的样本,设计新的64个碱基对探针(参见章节1
‑
1:材料,见上文)。记录实时rls光谱并且如上文所描述,分析500
‑
650纳米波长范围内的峰位置。
[0074]1‑
8:个别np的兴趣场(field of interest,foi)的显示
[0075]
个别纳米粒子的等离子foi定义为等离子灵敏度对比折射率变化的有效空间,其中等式1 (参见章节1
‑
10:数据分析,见下文)适用于计算与λ
max
红移幅度呈正比例的分子浓
度。推测 foi为立方体(图12a)。利用感测系统的winspec软件在ccd图像中直接用8个像素描绘foi的二维区域(图9d),基于比例尺校准,其长度及宽度测量为3.36微米(图8b)。在光谱监测之前设定此二维区域的长度及宽度,并且保持用于所有检测。如图8c所示,foi的高度 (h)为纳米结构的直径(d
np
)及t的总和,其中t定义为可诱发峰红移的区域的阈值厚度。详细地说,lspr峰偏移展现周期接近p=λ
max
/2n的振荡行为,其中n为表面上涂层的折射率 (图8d)(瑞迪威斯.t(rindzevicius,t.)等人,《物理化学学报(j phys chem c)》,111,11806
‑
11810, 2007)。在周期的第一半中,光谱展现厚度增加小于p/2的红移。在本文中,p/2的阈值厚度为 t。超出阈值厚度,lspr光谱开始蓝移且随后展现周期性振荡。先前研究证实dna层(n
dna
) 的折射率为2且蛋白质及dna根据其诱导的折射率变化类似地表现(扎克,v.v.(thacker,v. v.)等人,《自然通讯》,5,3448,2014;刘国梁等人(liu,g.l.et al.),《自然纳米技术(naturenanotech)》,1,47
‑
52,2006;迪普莫.c.(di primo,c.)等人,《分析生物化学(anal biochem)》, 368,148
‑
155,2007)。因此,λ
max
为561纳米的au桥接np的t计算为70.1纳米。最终,foi 的体积(l
×
w
×
h=v
foi
)确定为3.36微米
×
3.36微米
×
0.0844微米=0.953立方微米。
[0076]1‑
9:估算每个np的探针平均负载数(n
*
)
[0077]
基于纳米粒子及dna足迹(footprint)的建模定量地预测n
*
(图10)。详细地说,通过粒子的表面积除以探针有效足迹的面积来计算n
*
。足迹定义为各探针在纳米粒子表面上占据的平均面积。进行若干假设以用于计算。将纳米粒子建模为两个由圆柱体桥接的球体;彼此具有最近距离的足迹建模为球体上的圆及圆柱体上的椭圆;不考虑两个球体在玻璃上的接触点;以及假定探针均匀地分布于粒子表面上。
[0078]
根据球体的直径,球体上的足迹面积(s
sphere
)索引为6平方纳米。利用a
sphere
=a
′
sphere
‑
a
comact
计算两个球体的面积(a
sphere
)。其中a
′
sphere
为两个分离球体的面积且a
comact
为球体与圆柱体之间的接触面积;因此,a
sphere
=2
×
4π(d
sphere
/2)2‑2×
π(d
bridge
/2)2=1178平方纳米,且因此可装填于球体上的探针数为n
*sphere
=a
sphere
/s
spher
e=196。
[0079]
利用等式:n
*cylinder
=n
*short
‑
axis
×
n
*long
‑
axis
计算圆柱体外壁上的足迹面积,其中n
*short
‑
axis
为围绕圆周的足迹数量且n
*long
‑
axis
为圆柱体的轴下方的数量。然而,桥的长度(l
bridge
=2.39纳米) 并不允许沿圆柱体的轴负载超过一行探针,这是因为非弯曲表面上的两个行将具有长于2.39 纳米的足迹间隔距离(4.72纳米;海尔,h.d.(hill,h.d.)等人,《美国化学学会
·
纳米(acsnano)》,3,418
‑
424,2009)。因此,n
*cylinder
=n
*short
‑
axis
×
1=πd
bridge
/l
short
‑
axis
,其中d
bridge
为圆周长度且l
short
‑
axis
为由l
short
‑
axis
=2
×
√[(3.3618ln(d
bridge
/2)+0.1616)/π]得出的足迹的短轴长。n
*cylinder
测定为11,且最终n
*
=n
*sphere
+n
*cylinder
=207。
[0080]
由于粒子固定于平坦基板上,假设仅“有效负载边缘”线上方的表面可与dna有效地结合,其覆盖粒子的总表面积的59.4%(图10)。在饱和条件下,全部探针均捕获靶标。使用确立的等式n
dna
=[dna]v
foi
n
a
,可由[dna]
saturation
=59.4%n
*
/v
foi
/n
a
=215纳摩尔浓度来预测靶标 dna的饱和浓度([dna]
saturation
)。
[0081]1‑
10:数据分析
[0082]
np表面上对应于各分子结合步骤的ri变化表示为lsprλ
max
偏移(δλ
max
)。
[0083]
δλ
max
=m(δn)[1
‑
exp(
‑
2d/l
d
)]
ꢀꢀꢀꢀ
(1)
[0084]
其中m为折射率灵敏度,δn为由吸附物诱导的折射率变化,d为介电厚度且l
d
为电磁场衰减长度(近似为指数衰减;海斯,a.j.(haes,a.j.)等人,《美国化学学会杂志(jam chemsoc)》,124,10596
‑
10604,2002)。m、l
d
以及d为针对相同纳米粒子以及相同长度的探针及蛋白质的snps系统变量;且因此,δλ
max
与δn成正比例,δn与结合分析物的浓度成比例(斯塔洛夫,v.m.(starov,v.m.),《纳米科学:胶体及界面问题(nanoscience:colloidal and interracialaspects)》,crc出版,佛罗里达州波卡拉顿(boca raton,fl),2010)。基于δλ
max
的测量值,计算分析物浓度的变化。
[0085]
获得可靠δλ
max
的最低muts蛋白质浓度测定为如下snps程序的量化极限(limit of quantification,loq):
[0086]
loq=10σ/s
ꢀꢀꢀꢀ
(2)
[0087]
其中σ为信号的标准差且s为校准曲线的斜率。根据回归线的y截距的标准差估算σ的值。
[0088]
如下测定snps系统针对dna靶标的检测极限(lod):
[0089]
lod=3.3σ/s
ꢀꢀꢀꢀ
(3)
[0090]
信噪比(s/n)定义为δλ
max
的平均值(μ)与标准差的比率。在100%确定性下区分信号的s/n阈值为5(布什伯格,j.t.(bushberg,j.t.)等人,利平科特威廉姆斯及维尔金斯出版社 (lippincott williams&wilkins),宾夕法尼亚州费城(philadelphia,pa),2012)。
[0091]
s/n=μ/σ
ꢀꢀꢀꢀ
(4)
[0092]
在蛋白质核酸结合反应中,muts结合dna,从而形成msdna络合物。缔合为涉及两个反应物的二阶反应。
[0093][0094]
在概念上,结合及解离反应均涉及直接结合。在单dna链的层级下,muts缔合及解离为随机过程。通过简单的估算,au桥接np上的全部dna链可平均地用于结合。所使用的dna链长度指示以1∶1化学计量与muts结合;通过单指数过程描述结合的时程。在稳定状态下,结合速率等于释放速率:
[0095]
k
binding
[muts][dna]=k
dissociation
[msdna]
ꢀꢀꢀꢀ
(6)
[0096]
其中[muts]及[dna]分别为muts及dna的游离摩尔浓度;以且k
binding
及k
dissociation
分别为缔合(association)速率常数及解离(dissociation)速率常数。
[0097]
达到稳定状态之前,msdna络合物浓度的变化率等于其形成速率与解离速率之间的差。
[0098]
d[msdna]/dt=k
binding
[muts][dna]
‑
k
dissociation
[msdna](7)
[0099]
结合因尚未消耗反应物而以最大速率开始,且随后因反应物被消耗而减缓。反应随时间推移的程度可表示如下:
[0100][0101]
msdna的起始浓度([msdna
tn
])为零且因此可将以上等式转换成以下等式:
[0102][0103]
其中k
reaction
=k
binding
[muts]+k
dissociation
为观测的反应速率常数。k
dissociation
(测量muts与dna 解离的速度)与k
binding
(测量muts与dna结合的速度)的比率得到muts蛋白质的
平衡常数 (k
d
,以nm为单位),其用以评估双分子相互作用的强度且用以下等式来计算:
[0104][0105]
等式(9)及等式(10)的进一步转化可得到等式:
[0106][0107]
其中k
dissociation
与浓度无关且指示络合物在时间单位内自发分裂的概率(波拉德,t.d. (pollard,t.d.)等人,《分子生物细胞(mol biol cell)》24,1103
‑
1110,2013)。
[0108]
基于λ
max
变化的时程,由反应半衰期(τ
1/2
)评估达到最大δλ
max
的一半的结合时间:
[0109]
τ
1/2
=ln2/k
reaction
ꢀꢀꢀ
(12)
[0110]
实例2:构建的单纳米粒子感测平台的表征及点突变的检测
[0111]2‑
1:具有数字模拟的纳米粒子设计
[0112]
因为每个np在snps平台中充当信号转换器,所以np结构及形状应为均质的且可控的。这不包含形状不规则的纳米晶体(例如,分支链纳米星),因为基于合成原理,其形成具有实验性而不具有科学性。此外,多面体纳米结构的可控性会因缺乏可专门调谐目标晶体晶面的化学物质而受到限制且因此以相对较高产率产生np。因此,选择呈球体形状及棒形状的纳米结构作为用于snps的基板,因为可以均匀且可扩展的方式合成这两种形状的纳米结构。另外还引入诱导不同光谱响应并影响等离子耦合、偏光方向、信号强度以及ri灵敏度的量值的由纳米桥组成的结构以探索更高ri灵敏度(图11a)。金属np的灵敏度为测定生物/化学传感器的效用的主要因素。执行具有预先设计结构(其中周围介质的ri设定为改变)的单np的光学模拟,而不是使用络合物生物标记来定量ri灵敏度。对通过不同ri溶液诱导的单au
‑
np 的lspr波长(λ
max
)变化的分析证实在量化ri灵敏度方面为有效且简单的(特龙,p.l.,马, x.以及希姆,s.j.《纳米尺寸(nanoscale)》6,2307
‑
2315(2014))。观测到对应于各ri变化的λ
max
变化并以线性拟合形式表示(图11b),其中线的斜率表示np的ri灵敏度。金纳米棒 (aunr)展示比纳米球(aunsph)高的灵敏度(特龙,p.l.,马,x.以及希姆,s.j.《纳米尺寸》6,2307
‑
2315(2014))。纳米桥(au桥接np)结构展示比纳米棒更佳的效能,所述纳米棒先前被认为是最佳的结构。具体来说,au桥接np展示比aunr高两倍的灵敏度,使成为用于snps制造的超灵敏候选材料。这是由纳米间隙及纳米桥结构提供的高场浓度的直接结果。此外,等离子灵敏度与结构纵横比(ar)成比例地增加。然而,由于在结合超出rls光谱仪的波长范围的生物材料之后,较大ar可诱导大于800纳米的λ
max
,所以将au桥接np 的ar设定为2。
[0113]2‑
2:np的设计合成(svnthesis
‑
by
‑
design)
[0114]
研究使用双链dna(dsdna)进行金np的“方向特定”合成的可行性,固定于两个au 纳米晶种(auns;直径为约5纳米)之间的一个dsdna(长度为约3纳米)充当金原子结晶的方向特定引导(图2)。有趣的是,通过调节ph改变dsdna的表面电荷也会产生小于1纳米的纳米间隙(图11c)。在ph 5下,dsdna略微带负电并静电地聚集nh3oh
+
。反应物对 aucl4‑
及nh3oh
+
有可能彼此相遇,从而诱导沿dna进行的金结晶。在从auns
‑
dsdna边界到dsdna链中点的特定方向中出现结晶,其中纳米尺寸可控性是由dsdna长度限定。这一方法基本上与涉及使dna金属化或dna折纸术(dna origami)的现有途径不同,在所述现有途径中,形成连续的项
链或连续的凸出而结构精密度控制不佳(>100纳米)。因此,形成两个球体端长度为31.15
±
1.00纳米及直径为14.38
±
0.58纳米以及桥为8.79
±
0.96纳米的au 桥接np(表3)。
[0115]
[表3]
[0116][0117]
[a]
尺寸偏差为标准差与平均尺寸的比率。
[b]
从8批合成中获得纳米粒子并对tem图像平面中的194个粒子进行分析。
[0118]
所需形态的产率为87%,且纳米结构呈相对高的单分散性(图12)。相比之下,在ph 4 下,dsdna由于其pi为4
‑
4.5而带正电(郭,z.l.(guo,z.l.)等人,《软物质(soft matter)》,12,6669
‑
6674,2016)。dna因静电斥力排斥nh3oh
+
,且因此nh3oh
+
与aucl4‑
之间的反应主要出现在auns附近,其进一步自动催化围绕其表面的au原子的结晶。反应以au离子完全氧化成原子而结束,在两个纳米球(直径为17.01
±
1.07纳米)之间留下0.44纳米的间隙。
[0119]
在从auns
‑
dsdna边界到dsdna链中点的特定方向中出现结晶,其中纳米尺寸可控性是由dsdna长度限定。这一方法基本上与涉及使dna金属化或dna折纸术的现有途径不同,在所述现有途径中,形成连续的项链或连续的凸出而结构精密度控制不佳(>100纳米)。通过 x射线衍射及高分辨率穿透式电子显微镜术(hr
‑
tem)评估dna在合成au桥接np中的定向作用(图11d及e)。auns展现金(jcpds编号03
‑
0921)的fcc结构在38
°
(111)及44
°ꢀ
(200)处的峰。峰位置在dna定向结晶之后展现明显的偏移,表明dna在au桥接纳米结构中诱导显著的晶格应变。峰的狭窄线宽间接反映增大的粒子尺寸。此外,纳米尺寸桥体系的hr
‑
tem图像显示间隔为0.208
±
0.004纳米的晶体平面,与<100>结晶方向中的(200)晶格条纹相对应(马,x.,等人.《自然通讯》7,(2016))。综上所述,dna实现纳米尺寸桥及间隙的方向特定合成,所述纳米尺寸桥及间隙可用于灵敏的纳米等离子感测及成像。
[0120]2‑
3:具有au桥接np的snps
[0121]
研究在白光源下snps对单au桥接np的共振rls响应(图3)。在配备有白光源、暗场聚光器、100
×
物镜以及相机的暗场显微镜上观测岛个别np的光散射。白光照射使得有可能区分具有不同光学共振的个别np的光散射;其不会诱导使靶生物分子变性或干扰分子相互作用的较强能量及热量。使用暗场配置,在暗背景下明亮地照射光散射对象。aunp对lspr的刺激在很大程度上提高光散射,足以用裸眼识别并通过成像进行分析。np稀疏地分散于经设计微流体反应腔室的玻璃基板上。为测量单np的rls光谱,将高度灵敏的电荷耦合装置(ccd) 及瑞利光谱仪连接到显微镜上。随光散射强度与光谱波长的变化,用ccd记录单np的散射单色光。用洛伦兹演算法拟合散射光谱以消除来自周围光的噪音。
[0122]
光在np下产生的lspr充分增强光散射以允许直接观测到个别np;在另一方面,白光照射避免在微流体反应腔室中可使靶生物分子变性或阻断分子相互作用的高能量及热量(图 9a)。单au桥接np的rls光谱具有两个表面等离子峰(图9b):一个与横向方向上的电子振动相关,产生相对弱的共振带,约500纳米(类似于aunsph),而另一个与纵向方向上的
电子振动相关,约500纳米(类似于aunr)。本发明人关注纵向表面等离子峰,这是因为纵向模式比横向模式对介质的介电常数变化更灵敏(特龙,p.l.,马,x.以及希姆,s.j.《纳米尺寸》6,2307
‑
2315(2014))。介质的吸附物为ssdna及muts蛋白质。探针ssdna通过硫醇修饰固定于au桥接np并随后通过氢(h
‑
)键合及π
‑
π堆积而与靶ssdna杂交,在所述过程期间,λ
max
从561.1
±
0.5纳米红移到570.7
±
0.5纳米(步骤1及2;图9c)。在形成dna双螺旋之后,在λ
max
处观测到约9纳米的红移。之后,具有带正电表面序列的muts蛋白质独立地接触带负电的dna主链。dna中存在错配使得螺旋更复杂并诱导muts与保守性phe
‑
xaa
‑
glu 基序的特异性h
‑
结合,导致相对于裸np的λ
max
达到585.3
±
0.5纳米的红移高达24纳米(步骤2及3;图9c)。根据米氏理论(mie theory),金属np的lspr取决于局部介电环境的形状、尺寸以及ri。使用个别np消除形状及尺寸的差异;因此,lsprλ
max
偏移是归因于np 界面在dna杂交及后续muts结合之后的变化(图9d)。为验证在由muts识别突变的情况下出现的峰偏移的特异性,进行类似分析以测试人类血清中没有muts的dna靶标并且进行分析以测试没有突变的dna(即,完全匹配的靶标)。正如预期,在两个分析中观测到极少的光谱变化(图4)。在不移动微流体腔室的情况下引入含有muts的血清溶液及在95℃下用清洗缓冲液进行后续热处理之后,重复检测。相同地,观测到另一显著的红移,从而证实所制造的snps保存了muts针对dna突变的特异性。
[0123]2‑
4:感测的灵敏度
[0124]
根据两个参数研究snps感测方法的灵敏度:实现在特定检测时间内有效的lsprλ
max
偏移(δλ
max
)的muts蛋白质最低浓度(lod);及达到lod所需的检测时间。在muts溶液到达微流体腔室中的dna修饰的au桥接np之后,允许反应继续1分钟,之后在10秒内获得 rls光谱。添加过量的dna靶标以保证与探针的完全杂交。lspr读数的muts蛋白质有效浓度为6.17纳摩尔浓度,与10
‑
25纳摩尔浓度muts的线性范围内λ
max
的3.40纳米红移相对应(图9e)。随后,使用空白样本及一系列不同浓度的dna靶标进行测量以测定lod(图9f)。观测到5
‑
150纳摩尔浓度的分析范围,其中浓度与响应的曲线以r2为0.9954进行线性变化,超出所述分析范围,线性度不一致。在监测5纳摩尔浓度靶标时s/n为9.86。lod计算为8.63 纳摩尔浓度,其与用无标记qcm方法(苏,x.d.,罗贝勒,r.(robelek,r.),吴,y.j.(wu, y.j.),王,g.y.(wang,g.y.)以及纳尔,w.(knoll,w.),《分析化学(anal.chem.)》76,489
‑
494 (2004))获得的相当,且比由无标记spr批量检测(高特,m.(gotoh,m.)等人.《基因分析
‑
生物分子工程(genet anal
‑
biomol e)》14,47
‑
50(1997))测定的值的低数十倍。在1微升 /分钟的流速下达到此高灵敏度,且各检测所需的总样本体积为30微升的痕量样本。在对粒子感测无物理及化学干扰的情况下,容易从微流体腔室中洗涤出过量及非特异性的材料。相比之下,荧光感测方法需要大量的试剂及许多处理步骤(例如,muts在缓冲液中预浓度>3微摩尔浓度时需要持久的荧光团修饰,以获得<55%的标记效能)。凝胶迁移率偏移分析需要仅小体积的负载量,但样本必须高度浓缩以用于观测。muts足迹为24个碱基对,而蛋白质与dna 之间的交互分布于较大表面积上(约50个碱基对)所使用的51个碱基对ssdna探针确认点突变诱导信号变化与非特异性结合诱导变化之间的明显差异以最小化基于特异性靶结合的信号丢失。
[0125]2‑
5:单点突变的鉴别
[0126]
进行设计以鉴别brca1中的八个不同点突变。brca1基因突变包含乳癌(世界上最
常见的女性癌症)的最重要遗传易感性。约12%的女性将在其生命中患上乳癌,而因brca1突变带来的风险最高(59
‑
87%)。除极少的共同突变之外,brca1突变的光谱在不同种群中是异质的。选择brca1基因的八个多态性,包含在世界范围内最常见的单核苷酸取代(gt、gg、 ac、tc、aa以及ga)、插入(+c)以及缺失(
‑
c)。dna序列、突变名称、基因组位置、功能性结果以及靶标群概述于表1中。推测muts与点突变的序列特异性结合会改变不同的 lspr信号。另外,检测muts针对不同核苷酸变体的相对活性。在将snps平台注入到感测腔室中后,允许muts结合至dna共轭的au桥接np持续150秒,且每1秒监测单np的光学响应的变化(图13a)。根据实时信号响应,将muts负载于同源双链体(完全匹配)dna 上持续约15秒。这与muts形成短短暂的钳位并通过一维扩散滑动沿同源双链体dna移动的先前报告(吉隆,c.(jeong,c.)等人.《自然结构分子生物学(nat.struct.mol.biol.)》18, 379
‑
u174(2011))一致。错配鉴别会导致比同源双链体的结合时间长10倍的muts结合时间并诱导系列δλ
max
。因为这是由muts结合于dna共轭np后的ri变化所引起,所以时程清晰地反映muts在识别不同点突变中的不同活性。不同核苷酸变体会改变muts与dna之间的接触。举例来说,错配的胸腺嘧啶在n3位置处与muts的谷氨酸形成氢键,而错配的嘌呤在n7位置处形成氢键。muts中的氨基酸(例如苯丙氨酸)非特异性地堆积在错配碱基周围。这类不同特异性及非特异性相互作用导致可变的反应常数,其可通过muts
‑
dna相互作用的动力学分析来测定。
[0127]
muts与突变dna的相对活性r
act
)定义为muts结合至突变dna的效能,表示为r
act
=k
×
k
reaction
,其中k为占用常数(occupancy constant)且k
reaction
为蛋白质
‑
dna相互作用的速率常数。这是随机结合事件的简单估算,其中au桥接np上的dna可平均地用于muts;因此,相同检测条件准许相同的k。因此,可根据k
reaction
对r
act
进行求值。所使用的dna探针长度(51个碱基对)暗示与muts的1∶1结合化学计量;因此,结合及解离的时程可描述为单指数过程。通过与指数等式拟合,结合于不同dna靶标的muts针对点突变gt、gg、+c、 aa、tc、
‑
c、ac以及ga的k
reaction
(10
‑2s
‑
1)值分别为9.95
±
0.420、6.15
±
0.208、5.80
±
0.189、 4.92
±
0.214、3.82
±
0.212、3.60
±
0.243、3.25
±
0.184以及2.82
±
0.197。通过重新绘制随各靶标dna而变化的k
reacnon
值,muts针对突变的相对活性排序测定为gt>gg>+c>aa>tc>
ꢀ‑
c>ac>ga(图13b),其与先前凝胶迁移率偏移分析数据(德瑞克,v.c.(derocco,v.c.),萨斯,l.e.(sass,l.e.),秋,r.y.(qiu,r.y.),威尔,k.r.(weninger,k.r.)以及艾尔 d.a.(erie,d.a.),《生物化学(biochemistry
‑
us)》53,2043
‑
2052(2014))一致。点突变划分成四种类型:高度可鉴别的gt(k
reaction
>0.07)、可鉴别的gg、+c以及aa(k
reaction
=0.05
‑
0.07)、高度可检测的tc、
‑
c以及ac(k
reaction
=0.03
‑
0.05)以及可检测的ga(k
reacnon
<0.03)。
[0128]2‑
6:感测的可靠性
[0129]
已最清楚地证实结合于gt错配的muts的晶体结构及相互作用(格罗苏伊森,f.s. (groothuizen,f.s.)等人.elife 4,(2015))。因此,基于对muts与gt突变dna相互作用以形成名为“msdna”的络合物的k
reaction
的另一分析评估snps平台的可靠性。反应为涉及两个反应物的二阶反应:muts结合于dna并与解离的动力学可描述为 k
reacton
=k
binding
[muts]+k
dissociation
,其中k
binding
及k
dissociation
分别为结合速率常数及解离速率常数,且[muts]表示muts的游离摩尔浓度(图14a)。[muts]明显地影响反应动力且对应于δλ
max
的变化幅度。较高[muts]诱导平衡前λ
max
的较大增加,最终导致在相互作用结束时的较长偏移,相互作用因尚未消耗muts而以最大速率开始,之后反应以可预测方式减缓以达
到muts的平衡分布。通过指数拟合来定量地估算速率常数k
reaction
。重新绘制随[muts]而变化的k
reaction
,得到其中k
dissociation
为y截距且k
binding
为斜率的线性等式(图14b)。k
binding
为2.97
×
106m
‑1s
‑1,接近先前报告的批量动力学测量的3
‑6×
106m
‑1s
‑1(秋,r.y.等人.《美国科学院院报(proc.natlacad.sci.usa)》112,10914
‑
10919(2015))。对k
dissociation
与k
binding
之间的比率的动力学研究揭露muts与dna的解离平衡常数,即,k
d
,配体亲和力的基本参数。发现muts的k
d
为4.46 纳摩尔浓度,其与报告的smfret及批量测量2
‑
20纳摩尔浓度一致(吉隆,c.等人,《自然结构分子生物学》,18,379
‑
385,2011;杨,y.(yang,y.)等人,《核酸研究(nucleic acids res)》, 33,4322
‑
4334,2005)。这是通过对单np周围精确比例的muts平衡常数的动力学研究进行的验证,并支持snps分析用于诊断应用的效用。
[0130]2‑
7:用于临床诊断的针对点突变的muts亲和力图谱
[0131]
另外建立针对具有四种类型点突变的dna的蛋白质结合亲和力图谱(图15):高度可鉴别的((k
reaction
>0.07)、可鉴别的(k
reaction
=0.05
‑
0.07)、高度可检测的(k
reaction
=0.03
‑
0.05)以及可检测的(k
reaction
<0.03)。图谱示出针对各靶标、突变型以及诊断信号进行相对活性与反应半衰期的低输入、高保真性snps而获得的综合信息。具体来说,每个圆表示单点突变,其中直径及颜色分别反映用于量化lspr峰偏移及突变类别的信号响应。举例来说,具有gt突变的靶标dna产生14.2纳米的峰偏移,其值受靶标浓度(100纳摩尔浓度)的影响并与其成比例。突变类别在生物学上划分成三个类型,以蓝色、绿色以及红色进行标色,其中的蓝色类型指示转换突变(用嘧啶替换嘧啶或反过来也如此),绿色类型指示颠换突变(用嘌呤替换嘧啶或反过来也如此),且红色类型指示凸出突变(碱基插入或缺失)。每个圆的中心的y及x 坐标分别表示反应的相对活性及半衰期。由k
reaction
预测的相对活性视检测中使用的muts浓度及muts与靶标dna的k
d
而定。因此,可使用具有相同品质及数量的muts通过这种snps 方法鉴别出单点突变。反应的半衰期得以了解muts结合于个别np以达到最大lspr偏移的一半所花费的时间。这种了解可帮助说明例如嘌呤
‑
嘌呤突变在细胞中为何会展现比嘧啶
‑
嘧啶突变更佳的修复速率,如通过观测到相比于嘧啶
‑
嘧啶突变(例如,高度可检测的tc),muts 更强烈地结合至嘌呤
‑
嘌呤突变(可鉴别的gg及aa)所证实。这些结果也指示ac及ga突变的修复将不太有效,因为muts针对这些突变的相对活性比针对tc的相对活性更低。
[0132]
作为图谱的临床应用的原理证明(proof
‑
of
‑
principle),生物dna样本是由人类乳癌细胞系hcc1937及mcf7制备,分别作为分析物及对照(elstrodt,f等人,《癌症研究(cancer res)》, 66,41
‑
45,2006),并检测图谱中所示的八个突变中的潜在点突变的存在及类型(图5
‑
图7)。明显地,设计用于检测5382insc的玻璃芯片(表1)展现出一系列峰偏移,而其它芯片并未展现明显的信号变化(图15)。关于峰偏移响应于检测时间的动力学研究得到k
reaction
为5.73
±ꢀ
0.071(10
‑2s
‑1),其非常接近于图谱中+c的位置(图15及图17),从而证实hcc1937的brca1 含有单胞嘧啶复制。这个结果是由dna测序进行证实(图18)。另外,靶标的圆直径输出其浓度信息;例如根据图谱中用100纳摩尔浓度的标准浓度获得的+c的圆直径(12纳米),8.25 纳米直径指示68.8纳摩尔浓度的靶标。
[0133]
最终,此snps系统适用于检测用户指定基因组区域中的潜在点突变。位于染色体17的长臂的区域2条带1上的43047665处的潜在brca1点突变被指定用于判断卵巢癌细胞系 snu251。用相同的au桥接np但用新的64个碱基对探针来制造芯片。有趣的是,观测到光谱峰
的连续偏移,从而验证具有新探针的snps监测基因中的特定间隔的有效性(图19)。此外,诊断结果输入到图谱中指示突变类型高度类似于ac点突变。尽管新设计的探针与先前使用的ac探针的侧接序列不同,但k
reaction
(10
‑2s
‑1)得到类似值(检测中的3.20对比图谱中的 3.24)。紫色圆的较小直径指示诊断后靶标的低浓度。通过dna测序证实ac突变的预测是准确的(图20)。
[0134]
虽然已详细地描述本公开的细节,但将对本领域的技术人员显而易见的是这类细节仅仅是优选实施例且并不意图限制本发明的范围。因此,通过所附权利要求和其等效物来限定本发明的真实范围。
[0135]
工业实用性
[0136]
本发明的单纳米粒子生物传感器平台可用于不仅以高灵敏度及可靠性检测靶标,而且直接鉴别各种突变,从而实现突变的有效诊断。因此,本发明的单纳米粒子生物传感器平台可用于广泛范围的领域中,包含生物医学诊断。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1