黄原酸化合物分散体、共轭二烯系聚合物胶乳组合物和膜成型体的制作方法
1.本发明涉及黄原酸化合物分散体、共轭二烯系聚合物胶乳组合物和浸渍成型体等膜成型体,进一步详细而言,本发明涉及如下的黄原酸化合物分散体、使用这样的黄原酸化合物分散体得到的共轭二烯系聚合物胶乳组合物以及使用这样的共轭二烯系聚合物胶乳组合物得到的浸渍成型体等膜成型体,上述黄原酸化合物分散体在用作共轭二烯系聚合物等聚合物的硫化促进剂、制成浸渍成型体等膜成型体的情况下,除能够抑制速发型过敏(type i)症状的产生以外,还能够抑制迟发型过敏(type iv)症状的产生,而且使得到的浸渍成型体等膜成型体的撕裂强度高、撕裂强度的稳定性优异,而且可有效抑制针孔(pinhole)的产生。
背景技术:
2.一直以来,已知可将含有天然橡胶的胶乳的胶乳组合物浸渍成型,得到奶嘴、气球、手套、气囊、袋子等与人体接触而使用的浸渍成型体。然而,天然橡胶的胶乳含有会使人体引起速发型过敏(type i)症状的蛋白质,因此,作为与生物体粘膜或脏器直接接触的浸渍成型体,有时会存在问题。因此,正在研究使用合成橡胶的胶乳而不是天然橡胶的胶乳。
3.例如,专利文献1公开了一种在作为合成橡胶的合成聚异戊二烯的胶乳中添加氧化锌、硫和硫化促进剂而成的胶乳组合物作为浸渍成型用组合物。然而,在该专利文献1的技术中,虽然能够防止来自天然橡胶的蛋白质导致的速发型过敏(type i)的产生,但另一方面,在制成浸渍成型体的情况下,由于浸渍成型体所包含的硫化促进剂,在与人体接触时有时会引起迟发型过敏(type iv)的过敏症状。
4.现有技术文献
5.专利文献
6.专利文献1:国际公开第2014/129547号。
技术实现要素:
7.发明要解决的问题
8.本发明是鉴于这样的实际情况而完成的,目的在于提供如下的黄原酸化合物分散体、以及使用这样的黄原酸化合物分散体得到的共轭二烯系聚合物胶乳组合物和使用这样的共轭二烯系聚合物胶乳组合物得到的浸渍成型体等膜成型体,上述黄原酸化合物分散体在用作共轭二烯系聚合物等聚合物的硫化促进剂、制成浸渍成型体等膜成型体的情况下,除能够抑制速发型过敏(type i)症状的产生以外,还能够抑制迟发型过敏(type iv)症状的产生,而且使得到的浸渍成型体等膜成型体的撕裂强度高、撕裂强度的稳定性优异,而且可有效抑制针孔的产生。
9.用于解决问题的方案
10.本发明人等为了解决上述问题进行了深入研究,结果发现,根据使体积平均粒径
为0.001~9μm的黄原酸化合物分散在水或醇中而成的黄原酸化合物分散体,能够解决上述问题,基于这样的见解完成了本发明。
11.即,根据本发明,可提供一种使黄原酸化合物分散在水或醇中而成的黄原酸化合物分散体,上述黄原酸化合物的体积平均粒径为0.001~9μm。
12.本发明的黄原酸化合物分散体优选还含有非离子系表面活性剂和/或非离子阴离子系表面活性剂。
13.在本发明的黄原酸化合物分散体中,优选上述非离子系表面活性剂和/或非离子阴离子系表面活性剂具有聚氧化烯结构。
14.在本发明的黄原酸化合物分散体中,优选上述黄原酸化合物的95%体积累积径(d95)为0.1~43μm。
15.此外,在本发明的黄原酸化合物分散体中,优选上述黄原酸化合物为黄原酸盐,更优选为黄原酸的金属盐,进一步优选为黄原酸的锌盐。
16.此外,根据本发明,可提供含有共轭二烯系聚合物的胶乳、活化剂、和上述本发明的黄原酸化合物分散体的共轭二烯系聚合物胶乳组合物。
17.进而,根据本发明,可提供由这样的共轭二烯系聚合物胶乳组合物形成的膜成型体以及将这样的共轭二烯系聚合物胶乳组合物浸渍成型而成的浸渍成型体。
18.发明效果
19.根据本发明,能够提供如下的黄原酸化合物分散体、以及使用这样的黄原酸化合物分散体得到的共轭二烯系聚合物胶乳组合物和使用这样的共轭二烯系聚合物胶乳组合物得到的膜成型体,上述黄原酸化合物分散体在用作共轭二烯系聚合物等聚合物的硫化促进剂、制成浸渍成型体等膜成型体的情况下,除能够抑制速发型过敏(type i)症状的产生以外,还能够抑制迟发型过敏(type iv)症状的产生,而且使得到的浸渍成型体等膜成型体的撕裂强度高、撕裂强度的稳定性优异,而且可有效抑制针孔的产生。
具体实施方式
20.<黄原酸化合物分散体>
21.本发明的黄原酸化合物分散体是黄原酸化合物分散在水或醇中而成的,上述黄原酸化合物的体积平均粒径为0.001~9μm的范围。
22.作为本发明使用的黄原酸化合物,没有特别限定,可举出例如黄原酸、黄原酸盐等。
23.作为黄原酸盐,只要是具有黄原酸结构的盐化合物即可,没有特别限定,优选为黄原酸的金属盐,其中,优选通式(roc(=s)s)
x
‑
z(在此,r为直链状或支链状的烃,z为金属原子,x为与z的化合价一致的数,通常为1~4,优选为2~4,特别优选为2)所表示的化合物。此外,在黄原酸的金属盐中,更优选黄原酸的锌盐。
24.作为上述通式(roc(=s)s)
x
‑
z表示的黄原酸盐,没有特别限定,可举出例如二甲基黄原酸锌、二乙基黄原酸锌、二丙基黄原酸锌、二异丙基黄原酸锌、二丁基黄原酸锌、二戊基黄原酸锌、二己基黄原酸锌、二庚基黄原酸锌、二辛基黄原酸锌、二(2
‑
乙基己基)黄原酸锌、二癸基黄原酸锌、双十二烷基黄原酸锌、二甲基黄原酸钾、乙基黄原酸钾、丙基黄原酸钾、异丙基黄原酸钾、丁基黄原酸钾、戊基黄原酸钾、己基黄原酸钾、庚基黄原酸钾、辛基黄
原酸钾、2
‑
乙基己基黄原酸钾、癸基黄原酸钾、十二烷基黄原酸钾、甲基黄原酸钠、乙基黄原酸钠、丙基黄原酸钠、异丙基黄原酸钠、丁基黄原酸钠、戊基黄原酸钠、己基黄原酸钠、庚基黄原酸钠、辛基黄原酸钠、2
‑
乙基己基黄原酸钠、癸基黄原酸钠、十二烷基黄原酸钠等。在这些中,可以是异丙基黄原酸盐类、丁基黄原酸盐类,优选上述通式(roc(=s)s)
x
‑
z中的x为2以上的黄原酸盐,更优选二异丙基黄原酸盐类、二丁基黄原酸盐类,进一步优选二异丙基黄原酸锌、二丁基黄原酸锌,特别优选二异丙基黄原酸锌。这些黄原酸盐可以单独使用一种,也可以并用多种。
25.另外,这些黄原酸化合物可以单独使用一种,也可以组合使用两种以上。
26.本发明使用的黄原酸化合物是作为硫化促进剂发挥作用的化合物,因此,本发明的黄原酸化合物分散体能够优选地用作共轭二烯系聚合物等聚合物的硫化促进剂。黄原酸化合物在硫化时作为硫化促进剂发挥作用,在进行硫化后,通过硫化时施加的热等分解为醇和二硫化碳等。此外,通过分解生成的醇和二硫化碳等由于硫化时施加的热等通常会挥发,因此能够抑制黄原酸化合物在得到的共轭二烯系聚合物等聚合物的硫化物中的残留量。因此,根据本发明,不使用一直以来成为迟发型过敏(type iv)症状的产生原因的硫化促进剂(例如,秋兰姆系硫化促进剂、二硫代氨基甲酸盐系硫化促进剂、噻唑系硫化促进剂等),而使用本发明的黄原酸化合物分散体,由此能够减少在得到的共轭二烯系聚合物等聚合物的硫化物(例如浸渍成型体等膜成型体)中的引起过敏的物质的含量,因此除能够抑制速发型过敏(type i)症状的产生之外,还能够抑制迟发型过敏(type iv)症状的产生。
27.此外,在本发明中,在黄原酸化合物分散体中,使黄原酸化合物以颗粒状或粉末状分散,且使黄原酸化合物分散体中分散的黄原酸化合物的体积平均粒径在0.001~9μm的范围。根据本发明,通过使黄原酸化合物分散体中分散的黄原酸化合物的体积平均粒径在0.001~9μm的范围,在将本发明的黄原酸化合物分散体用作共轭二烯系聚合物等聚合物的硫化促进剂、制成浸渍成型体等膜成型体的情况下,能够使得到的浸渍成型体等膜成型体撕裂强度高、撕裂强度的稳定性优异,而且可有效抑制针孔的产生。
28.黄原酸化合物分散体中分散的黄原酸化合物的体积平均粒径为0.001~9μm的范围即可,优选在0.05~9μm的范围,更优选在0.05~7μm的范围,进一步优选在0.07~5μm的范围。当黄原酸化合物分散体中分散的黄原酸化合物的体积平均粒径过小时,难以在作为分散介质的水或醇中均匀地分散,因此有得不到期望的特性的风险。另一方面,当体积平均粒径过大时,得到的浸渍成型体等膜成型体的撕裂强度变低,撕裂强度的稳定性变差,且针孔的抑制效果变得不充分。此外,当体积平均粒径过大时,实现所期望的撕裂强度所需要的熟化(预硫化)时间变长,生产率变差。
29.此外,在本发明中,黄原酸化合物分散体中分散的黄原酸化合物的体积平均粒径为上述范围即可,优选黄原酸化合物的95%体积累积径(d95)在0.1~43μm的范围,更优选在0.1~40μm的范围,进一步优选在0.1~35μm的范围,特别优选在0.1~20μm的范围。通过使95%体积累积径(d95)在上述范围,能够进一步加强撕裂强度和撕裂强度的提高效果、以及针孔产生的抑制效果。另外,黄原酸化合物的体积平均粒径和95%体积累积径(d95)能够使用例如激光衍射散射式粒度分布计进行测定。
30.本发明的黄原酸化合物分散体中的黄原酸化合物的含有比例相对于黄原酸化合物分散体整体,优选为1~60重量%,更优选为10~50重量%,进一步优选为30~50重量%。
通过使黄原酸化合物的含有比例为上述范围,能够使黄原酸化合物分散体的保存稳定性更优异。
31.此外,本发明的黄原酸化合物分散体优选除含有上述的黄原酸化合物以外,还含有非离子系表面活性剂和/或非离子阴离子系表面活性剂。
32.根据本发明,通过使上述的黄原酸化合物与非离子系表面活性剂和/或非离子阴离子系表面活性剂一起分散在水或醇中,能够使黄原酸化合物更良好地分散,由此能够进一步提高黄原酸化合物的作为硫化促进剂的效果,由此,能够缩短在得到共轭二烯系聚合物等聚合物的硫化物时的硫化时间(特别是熟化(预硫化)所需要的时间),能够实现生产率的提高。另外,在本发明中,优选使用非离子系表面活性剂和非离子阴离子系表面活性剂中的至少一者,更优选使用非离子系表面活性剂。
33.作为非离子系表面活性剂,只要是非离子性的表面活性剂即可,没有特别限定,可举出例如聚氧化烯二醇、聚氧化烯烷基醚、聚氧化烯烷基苯基醚、聚氧乙烯苯乙烯化苯基醚、聚氧乙烯(固化)蓖麻油、聚氧乙烯烷基胺、脂肪酸烷醇酰胺等。
34.作为聚氧化烯二醇,可举出例如聚氧乙烯二醇、聚氧丙烯二醇、聚氧乙烯聚氧丙烯二醇等聚氧丙烯二醇环氧乙烷加成物等。
35.作为聚氧化烯烷基醚,可举出例如加成了1~50个(优选1~10个)环氧丙烷和/或环氧乙烷的直链状或支链状醚。在这些中,可举出加成了1~50个(优选1~10个)环氧丙烷的直链状或支链状醚、加成了1~50个(优选1~10个)环氧乙烷的直链状或支链状醚、进行嵌段或无规加成了合计2~50个(优选2~10个)环氧乙烷和环氧丙烷的直链状或支链状醚等,可举出聚氧乙烯十二烷基醚、聚氧乙烯月桂基醚等。
36.作为聚氧化烯烷基苯基醚,可举出对烷基苯酚加成1~50个(优选1~10个)环氧丙烷和/或环氧乙烷的化合物等。
37.作为聚氧乙烯苯乙烯化苯基醚,可举出(单、二、三)苯乙烯化苯酚的环氧乙烷加成物等,在这些中,优选作为二苯乙烯化苯酚的环氧乙烷加成物的聚氧乙烯二苯乙烯化苯基醚。
38.作为聚氧乙烯(固化)蓖麻油,可举出蓖麻油或固化蓖麻油的环氧乙烷加成物。
39.作为脂肪酸烷醇酰胺,可举出例如月桂酸二乙醇酰胺、棕榈酸二乙醇酰胺、十四酸二乙醇酰胺、硬脂酸二乙醇酰胺、油酸二乙醇酰胺、棕榈油脂肪酸二乙醇酰胺、椰子油脂肪酸二乙醇酰胺等。
40.在非离子系表面活性剂中,优选具有聚氧化烯结构的非离子系表面活性剂,更优选具有聚氧乙烯结构的非离子系表面活性剂,更优选为聚氧乙烯的烃基化醚,进一步优选聚氧化烯烷基醚和聚氧乙烯二苯乙烯化苯基醚,特别优选聚氧乙烯二苯乙烯化苯基醚。非离子系表面活性剂可以单独使用一种,也可以组合使用两种以上。
41.此外,作为非离子阴离子系表面活性剂,只要是阴离子性表面活性剂(即在水中解离为离子,阴离子部分显现表面活性的物质)且在其分子主链中具有作为非离子性的表面活性剂发挥作用的链段例如聚环氧烷链即可,没有特别限定。
42.作为这样的非离子阴离子系表面活性剂,可举出例如下述通式(1)所表示的化合物。
43.r1‑
o
‑
(cr2r3cr4r5)
n
‑
so3m
ꢀꢀ
(1)
44.(上述通式(1)中,r1表示碳原子数为6~16的烷基或可以被碳原子数为1~25的烷基取代的碳原子数为6~14的芳基,r2~r5为各自独立地选自氢或甲基中的基团,m为碱金属原子或铵离子,n为3~40。)
45.作为非离子阴离子系表面活性剂的具体例子,可举出聚氧乙烯月桂基醚硫酸盐、聚氧乙烯十六烷基醚硫酸盐、聚氧乙烯硬脂基醚硫酸盐、聚氧乙烯油基醚硫酸盐等聚氧乙烯烷基醚硫酸盐;聚氧乙烯壬基苯基醚硫酸盐、聚氧乙烯辛基苯基醚硫酸盐、聚氧乙烯二苯乙烯基醚硫酸盐等聚氧乙烯芳基醚硫酸盐等。
46.在非离子阴离子系表面活性剂中,优选具有聚氧化烯结构的非离子阴离子系表面活性剂,更优选具有聚氧乙烯结构的非离子阴离子系表面活性剂。非离子阴离子性表面活性剂可以单独使用一种,也可以组合使用两种以上。
47.本发明的黄原酸化合物分散体中的非离子系表面活性剂和/或非离子阴离子系表面活性剂的含量没有特别限定,相对于100重量份的黄原酸化合物,优选为0.1~30重量份,更优选为1~20重量份,进一步优选为4~15重量份,特别优选为5.5~9.5重量份。通过使非离子系表面活性剂和/或非离子阴离子系表面活性剂的含量为上述范围,能够进一步提高黄原酸化合物分散体中的黄原酸化合物的分散性,由此能够进一步提高得到共轭二烯系聚合物等聚合物的硫化物时的硫化时间的缩短效果。
48.作为本发明的黄原酸化合物分散体的制造方法,没有特别限定,优选将黄原酸化合物、根据需要使用的非离子系表面活性剂和/或非离子阴离子系表面活性剂、以及水或醇(例如选自甲醇、乙醇、丙醇和丁醇中的至少一种)混合、接下来对得到的混合液进行破碎处理的方法,特别优选通过调节破碎处理的条件,使黄原酸化合物的体积平均粒径为上述范围。在此,作为破碎处理,只要是能够使分散体中所含有的黄原酸化合物的破碎、缓和其凝聚的处理即可,没有特别限定,可举出例如使用利用了剪切作用、磨碎作用的破碎装置的方法,使用搅拌式的破碎装置的方法等使用公知的破碎装置的方法。具体而言,能够使用辊磨机、锤磨机、振动磨、喷射磨、球磨机、行星式球磨机、珠磨机、砂磨机、三辊磨机等破碎装置。在这些中,从能够优选地控制分散体中的黄原酸化合物的体积平均粒径的观点出发,优选使用球磨机、行星式球磨机或珠磨机进行破碎处理的方法。
49.例如,在使用球磨机进行破碎处理的情况下,优选以如下的条件进行破碎处理:作为介质,使用介质大小优选为更优选为的介质,转速优选为10~300rpm、更优选为10~100rpm,处理时间优选为24~120小时、更优选为24~72小时。此外,在使用行星式球磨机进行破碎处理的情况下,优选以如下的条件进行破碎处理:作为介质,使用介质大小优选质,使用介质大小优选更优选的介质,转速优选为100~1000rpm、更优选为100~500rpm,处理时间优选为0.25~5小时、更优选为0.25~3小时。进而,在使用珠磨机进行破碎处理的情况下,优选以如下的条件进行破碎处理:作为介质,使用介质大小优选更优选的介质,转速优选为1000~10000rpm、更优选为1000~5000rpm,处理时间优选为0.25~5小时、更优选为0.25~3小时。
50.<共轭二烯系聚合物胶乳组合物>
51.本发明的共轭二烯系聚合物胶乳组合物含有共轭二烯系聚合物的胶乳、硫化剂和上述的本发明的黄原酸化合物分散体。
52.作为构成共轭二烯系聚合物的胶乳的共轭二烯系聚合物,没有特别限定,可举出例如合成聚异戊二烯、苯乙烯
‑
异戊二烯
‑
苯乙烯嵌段共聚物(sis)、除去蛋白质的天然橡胶、含腈基共轭二烯系共聚物等。在这些中,优选合成异戊二烯、sis、除去蛋白质的天然橡胶等含有异戊二烯单元的聚合物,特别优选合成聚异戊二烯。另外,作为共轭二烯系聚合物,可以是通过具有羧基的单体进行改性得到的羧基改性共轭二烯系聚合物。
53.在使用合成聚异戊二烯作为共轭二烯系聚合物的情况下,合成聚异戊二烯可以是异戊二烯的均聚物,也可以是与能够与异戊二烯共聚的其他的烯属不饱和单体进行共聚的聚合物。从容易得到柔软且拉伸强度优异的浸渍成型体等膜成型体的方面出发,合成聚异戊二烯中的异戊二烯单元的含量相对于全部单体单元优选为70重量%以上、更优选为90重量%以上、进一步优选为95重量%以上、特别优选为100重量%(异戊二烯的均聚物)。
54.作为能够与异戊二烯共聚的其他的烯属不饱和单体,可举出例如丁二烯、氯丁二烯、1,3
‑
戊二烯等除异戊二烯以外的共轭二烯单体;丙烯腈、甲基丙烯腈、富马腈、α
‑
氯丙烯腈等烯属不饱和腈单体;苯乙烯、烷基苯乙烯等乙烯基芳香族单体;(甲基)丙烯酸甲酯(意思是“丙烯酸甲酯和/或甲基丙烯酸甲酯”,以下(甲基)丙烯酸乙酯等也同样)、(甲基)丙烯酸乙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸
‑2‑
乙基己酯等烯属不饱和羧酸酯单体等。这些能够与异戊二烯共聚的其他的烯属不饱和单体可以单独使用一种,也可以并用多种。
55.合成聚异戊二烯能够通过现有公知的方法,使用例如三烷基铝
‑
四氯化钛形成的齐格勒系聚合催化剂、正丁基锂、仲丁基锂等烷基锂聚合催化剂,在非活性聚合溶剂中将异戊二烯与根据需要而使用的能够共聚的其他的烯属不饱和单体进行溶液聚合来得到。通过溶液聚合得到的合成聚异戊二烯的聚合物溶液能够直接用于合成聚异戊二烯胶乳的制造,也能够从该聚合物溶液中取出固体的合成聚异戊二烯后,溶于有机溶剂,用于合成聚异戊二烯胶乳的制造。此外,在通过上述的方法得到合成聚异戊二烯的聚合物溶液的情况下,可以除去残留在聚合物溶液中的聚合催化剂的残渣等杂质。此外,可以在聚合中或聚合后的溶液中添加后述的抗老化剂。此外,还能够使用市售的固体的合成聚异戊二烯。
56.作为合成聚异戊二烯中的异戊二烯单元,根据异戊二烯的键状态,存在顺式键单元、反式键单元、1,2
‑
乙烯基键单元、3,4
‑
乙烯基键单元4类。从提高得到的浸渍成型体等膜成型体的拉伸强度的观点出发,合成聚异戊二烯所包含的异戊二烯单元中的顺式键单元的含有比例相对于全部异戊二烯单元优选为70重量%以上、更优选为90重量%以上、进一步优选为95重量%以上。
57.合成聚异戊二烯的重均分子量以凝胶渗透色谱分析的标准聚苯乙烯换算计,优选为10000~5000000、更优选为500000~5000000、进一步优选为800000~3000000。通过使合成聚异戊二烯的重均分子量为上述范围,会提高浸渍成型体等膜成型体的拉伸强度,并且合成聚异戊二烯胶乳有变得容易制造的倾向。
58.此外,合成聚异戊二烯的聚合物门尼粘度(ml1+4、100℃)选为50~85、更优选为60~85、进一步优选为70~85。
59.作为得到合成聚异戊二烯胶乳的方法,可举出例如(1)在阴离子性表面活性剂的存在下,将溶解或微分散于有机溶剂的合成聚异戊二烯的溶液或微悬浊液在水中进行乳化,根据需要除去有机溶剂,制造合成聚异戊二烯胶乳的方法;(2)在阴离子性表面活性剂的存在下,将异戊二烯单独或异戊二烯与能够与其共聚的烯属不饱和单体的混合物进行乳
液聚合或悬浮聚合,直接制造合成聚异戊二烯胶乳的方法,从能够使用异戊二烯单元中顺式键单元的比例高的合成聚异戊二烯、容易得到拉伸强度等机械特性优异的浸渍成型体等膜成型体的方面出发,优选上述制造方法(1)。
60.作为上述(1)的制造方法所使用的有机溶剂,能够举出例如苯、甲苯、二甲苯等芳香族烃溶剂;环戊烷、环戊烯、环己烷、环己烯等脂环族烃溶剂;戊烷、己烷、庚烷等脂肪族烃溶剂;二氯甲烷、氯仿、二氯乙烷等卤代烃溶剂等。在这些中,优选脂环族烃溶剂,特别优选环己烷。
61.另外,有机溶剂的使用量相对于100重量份的合成聚异戊二烯,优选为2000重量份以下、更优选为20~1500重量份、进一步优选为500~1500重量份。
62.作为上述(1)的制造方法使用的阴离子性表面活性剂,可举出例如月桂酸钠、肉豆蔻酸钾、棕榈酸钠、油酸钾、亚麻酸钠、松香酸钠等脂肪酸盐;十二烷基苯磺酸钠、十二烷基苯磺酸钾、癸基苯磺酸钠、癸基苯磺酸钾、十六烷基苯磺酸钠、十六烷基苯磺酸钾等烷基苯磺酸盐;二(2
‑
乙基己基)磺基琥珀酸钠、二(2
‑
乙基己基)磺基琥珀酸钾、二辛基磺基琥珀酸钠等烷基磺基琥珀酸盐;月桂基硫酸钠、月桂基硫酸钾等烷基硫酸酯盐;聚氧乙烯月桂基醚硫酸钠、聚氧乙烯月桂基醚硫酸钾等聚氧乙烯烷基醚硫酸酯盐;磷酸钠月桂酯、磷酸钾月桂酯等磷酸单烷基酯盐等。
63.在这些阴离子性表面活性剂中,优选脂肪酸盐、烷基苯磺酸盐、烷基磺基琥珀酸盐、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐,特别优选脂肪酸盐和烷基苯磺酸盐。
64.此外,由于在能够更高效地除去来自合成聚异戊二烯的微量残留的聚合催化剂(特别是铝和钛)、在制造共轭二烯系聚合物胶乳组合物时抑制凝聚物的产生,因此优选将选自烷基苯磺酸盐、烷基磺基琥珀酸盐、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐中的至少一种与脂肪酸盐并用,特别优选将烷基苯磺酸酯盐与脂肪酸盐并用。在此,作为脂肪酸盐,优选松香酸钠和松香酸钾,此外,作为烷基苯磺酸盐,优选十二烷基苯磺酸钠和十二烷基苯磺酸钾。此外,这些表面活性剂可以单独使用一种,也可以并用两种以上。
65.另外,如上所述,通过将选自烷基苯磺酸盐、烷基磺基琥珀酸盐、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐中的至少一种与脂肪酸盐并用,得到的胶乳含有选自烷基苯磺酸盐、烷基磺基琥珀酸盐酯、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐中的至少一种以及脂肪酸盐。
66.此外,在上述(1)的制造方法中,可以并用除阴离子性表面活性剂以外的表面活性剂,作为这样的除阴离子性表面活性剂以外的表面活性剂,可举出α,β
‑
不饱和羧酸的磺基酯、α,β
‑
不饱和羧酸的硫酸酯、磺基烷基芳基醚等共聚性的表面活性剂。
67.上述(1)的制造方法使用的阴离子性表面活性剂的使用量相对于100重量份的合成聚异戊二烯,优选为0.1~50重量份、更优选为0.5~30重量份。另外,在使用两种以上的表面活性剂的情况下,优选使这些的合计的使用量为上述范围。即,例如在并用选自烷基苯磺酸盐、烷基磺基琥珀酸盐、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐中的至少一种以及脂肪酸盐的情况下,优选使这些的使用量的合计为上述范围。当阴离子性表面活性剂的使用量过少时,乳化时有可能大量产生凝聚物,反之当过多时,可能会变得容易发泡,得到的浸渍成型体等膜成型体可能产生针孔。
68.此外,作为阴离子性表面活性剂,在并用选自烷基苯磺酸盐、烷基磺基琥珀酸盐、
烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐中的至少一种和脂肪酸盐的情况下,以“脂肪酸盐”∶“选自烷基苯磺酸盐、烷基磺基琥珀酸盐酯、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐的中的至少一种表面活性剂的合计”的重量比计,这些的使用比例优选为1∶1~10∶1的范围、更优选为1∶1~7∶1的范围。当选自烷基苯磺酸盐、烷基磺基琥珀酸盐、烷基硫酸酯盐和聚氧乙烯烷基醚硫酸酯盐中的至少一种表面活性剂的使用比例过多时,在处理合成聚异戊二烯时可能会变得剧烈发泡,由此需要长时间的静置、添加消泡剂等操作,有可能导致操作性变差和成本上升。
69.上述(1)的制造方法使用的水量相对于100重量份的合成聚异戊二烯的有机溶剂溶液优选为10~1000重量份、更优选为30~500重量份、最优选为50~100重量份。作为使用的水的种类,可举出硬水、软水、离子交换水、蒸馏水、沸石水等,优选软水、离子交换水和蒸馏水。
70.在阴离子性表面活性剂存在下,对于将溶解或微分散于有机溶剂的合成聚异戊二烯的溶液或微悬浊液在水中进行乳化的装置,一般只要是作为乳化机或分散机市售的装置,则能够没有特别限定地使用。作为对合成聚异戊二烯的溶液或微悬浊液添加阴离子性表面活性剂的方法,没有特别限定,可以预先在水中或者在合成聚异戊二烯的溶液或微悬浊液中的任一种中添加,或者预先向两者添加;可以在进行乳化操作的中途添加至乳化液,可以一次性添加、也可以分次添加。
71.作为乳化装置,可举出例如商品名“homogenizer”(ika公司制)、商品名“polytron”(kinematica公司制)、商品名“tk auto homomixer”(特殊机化工业公司制)等分批式乳化机;商品名“tk pipeline homomixer”(特殊机化工业公司制)、商品名“colloid mill”(shinko pantec公司制)、商品名“thrasher”(nippon coke&engineering公司制)、商品名“trigonal湿式微粉碎机”(三井三池化工机公司制)、商品名“cavitron”(eurotec公司制)、商品名“milder”(太平洋机工公司制)、商品名“fine flow mill”(太平洋机工公司制)等连续式乳化机;商品名“microfluidizer”(mizuho工业公司制)、商品名“nanomizer”(nanomizer公司制)、商品名“apv gaulin”(gaulin公司制)等高压乳化机;商品名“膜乳化机”(冷化工业公司制)等膜乳化机;商品名“vibromixer”(冷化工业公司制)等振动式乳化机;商品名“超声波homogenizer”(branson公司制)等超声波乳化机等。另外,通过乳化装置进行的乳化操作的条件没有特别限定,以成为期望的分散状态的方式,适当选择处理温度、处理时间等即可。
72.在上述(1)的制造方法中,优选从经过乳化操作而得到的乳化物中除去有机溶剂。作为从乳化物中除去有机溶剂的方法,优选能够使得到的合成聚异戊二烯胶乳中有机溶剂(优选为脂环族烃溶剂)的含量为500重量ppm以下的方法,能够采用例如减压蒸馏、常压蒸馏、水蒸气蒸馏、离心分离等方法。
73.在上述制造方法(1)中,优选从经过乳化操作而得到的乳化物中除去有机溶剂而得到合成聚异戊二烯胶乳。作为从乳化物中除去有机溶剂的方法,只要是能够使得到的合成聚异戊二烯胶乳中作为有机溶剂的脂环族烃溶剂和芳香族烃溶剂的合计含量为500重量ppm以下的方法,则没有特别限定,能够采用减压蒸馏、常压蒸馏、水蒸气蒸馏、离心分离等方法。
74.此外,为了在除去有机溶剂后,根据需要提高合成聚异戊二烯胶乳的固体成分浓
度,可以通过减压蒸馏、常压蒸馏、离心分离、膜浓缩等方法实施浓缩操作,特别地,从能够提高合成聚异戊二烯的固体成分的浓度,并且减少合成聚异戊二烯胶乳中的表面活性剂的残留量的观点出发,优选进行离心分离。
75.离心分离优选在如下的条件下实施:例如使用连续离心分离、离心力优选为100~10000g、离心分离前的合成聚异戊二烯胶乳的固体成分浓度优选为2~15重量%、送入离心分离机的流速优选为500~1700kg/hr、离心分离机的背压(表压)优选为0.03~1.6mpa,作为离心分离后的轻液,能够得到合成聚异戊二烯胶乳。而且,由此能够降低合成聚异戊二烯胶乳中表面活性剂的残留量。
76.合成聚异戊二烯胶乳的固体成分浓度优选为30~70重量%,更优选为40~70重量%。当固体成分浓度过低时,由于共轭二烯系胶乳组合物的固体成分浓度变低,所以得到的浸渍成型体等膜成型体的膜厚变薄,容易破裂。反之当固体成分浓度过高时,合成聚异戊二烯胶乳的粘度变高,有时通过配管的运送、在调配罐内的搅拌变得困难。
77.合成聚异戊二烯胶乳的体积平均粒径优选为0.1~10μm,更优选为0.5~3μm,进一步优选为0.5~2.0μm。通过使该体积平均粒径为上述范围,胶乳粘度变得适宜,变得容易处理,并且能够在贮藏合成聚异戊二烯胶乳时,抑制胶乳表面生成皮膜。
78.此外,可以对合成聚异戊二烯胶乳添加胶乳领域通常添加的ph调节剂、消泡剂、防腐剂、交联剂、螯合剂、除氧剂、分散剂、抗老化剂等添加剂。作为ph调节剂,可举出例如氢氧化钠、氢氧化钾等碱金属的氢氧化物;碳酸钠、碳酸钾等碱金属的碳酸盐;碳酸氢钠等碱金属的碳酸氢盐;氨;三甲胺、三乙醇胺等有机胺化合物等,优选碱金属的氢氧化物或氨。
79.此外,作为共轭二烯系聚合物,如上所述,能够使用苯乙烯
‑
异戊二烯
‑
苯乙烯嵌段共聚物(sis)。另外,在sis中,“s”表示苯乙烯嵌段,“i”表示异戊二烯嵌段。
80.sis能够通过现有公知的方法得到,例如将正丁基锂等活性有机金属作为引发剂,在非活性聚合溶剂中将异戊二烯和苯乙烯进行嵌段共聚来得到。而且,得到的sis共聚物溶液可以直接用于sis胶乳的制造,也能够从该聚合物溶液中取出固态的sis后,将该固态的sis溶于有机溶剂,用于sis胶乳的制造。作为sis胶乳的制造方法没有特别限定,优选将溶解或微分散于有机溶剂的sis的溶液或微悬浊液在表面活性剂的存在下在水中乳化,根据需要除去有机溶剂,制造sis胶乳的方法。此时,可以在合成后除去残留在聚合物溶液中的聚合催化剂的残渣等杂质。此外,可以在聚合中或聚合后的溶液中添加后述的抗老化剂。此外,也能够使用市售的固态的sis。
81.作为有机溶剂,能够使用与上述合成聚异戊二烯的情况相同的溶剂,优选芳香族烃溶剂和脂环族烃溶剂,特别优选环己烷和甲苯。另外,有机溶剂的使用量相对于100重量份的sis通常为50~2000重量份、优选为80~1000重量份、更优选为100~500重量份、进一步优选为150~300重量份。
82.作为表面活性剂,能够示例与上述合成聚异戊二烯的情况相同的表面活性剂,优选阴离子性表面活性剂,特别优选松香酸钠和十二烷基磺酸钠。
83.表面活性剂的使用量相对于100重量份的sis优选为0.1~50重量份,更优选为0.5~30重量份。当该量过少时,胶乳的稳定性有变差的倾向,反之当过多时,可能变得容易起泡、在浸渍成型时产生问题。
84.上述的sis胶乳的制造方法中使用的水量相对于100重量份的sis的有机溶剂溶
液,优选为10~1000重量份、更优选为30~500重量份、进一步优选为50~100重量份。作为使用的水的种类,可举出硬水、软水、离子交换水、蒸馏水、沸石水等。此外,可以用甲醇等醇为代表的极性溶剂与水并用。
85.作为单体的添加方法,能够示例与上述合成聚异戊二烯的情况相同的方法。此外,对于将sis的有机溶剂溶液或微悬浊液在表面活性剂存在下在水中乳化的装置,能够示例与上述合成聚异戊二烯的情况相同的装置。而且,表面活性剂的添加方法没有特别限定,可以预先在水中或者在sis的溶液或微悬浊液中的任一者中添加,或者预先向两者添加;可以在进行乳化操作时添加至乳化液,可以一次性添加,也可以分次添加。
86.在上述的sis胶乳的制造方法中,优选从经过乳化操作得到的乳化物中除去有机溶剂而得到sis胶乳。从乳化物中除去有机溶剂的方法没有特别限定,能够采用减压蒸馏、常压蒸馏、水蒸气蒸馏、离心分离等方法。
87.此外,为了在除去有机溶剂后根据需要提高sis胶乳的固体成分浓度,可以通过减压蒸馏、常压蒸馏、离心分离、膜浓缩等方法实施浓缩操作。
88.sis胶乳的固体成分浓度优选为30~70重量%,更优选为50~70重量%。当固体成分浓度过低时,由于共轭二烯系聚合物胶乳组合物的固体成分浓度变低,因此在制成浸渍成型体等膜成型体时,膜厚变薄,容易破裂。反之当固体成分浓度过高时,sis胶乳的粘度变高,通过配管的运送、在调配罐内的搅拌变得困难。
89.此外,可以对sis胶乳添加胶乳领域通常添加的ph调节剂、消泡剂、防腐剂、交联剂、螯合剂、除氧剂、分散剂、抗老化剂等添加剂。作为ph调节剂,能够示例与上述的合成聚异戊二烯的情况相同的ph调节剂,优选碱金属的氢氧化物或氨。
90.这样得到sis胶乳所包含的sis中的苯乙烯嵌段中的苯乙烯单元的含量相对于全部单体单元,优选为70~100重量%、更优选为90~100重量%、进一步优选为100重量%。此外,sis中的异戊二烯嵌段中的异戊二烯单元的含量相对于全部单体单元优选为70~100重量%、更优选为90~100重量%、进一步优选为100重量%。
91.另外,sis中的苯乙烯单元和异戊二烯单元的含有比例以“苯乙烯单元∶异戊二烯单元”的重量比计,通常为1∶99~90∶10的范围、优选为3∶97~70∶30的范围、更优选为5∶95~50∶50的范围、进一步优选为10∶90~30∶70的范围。
92.sis的重均分子量以凝胶渗透色谱分析的标准聚苯乙烯换算计,优选为10000~1000000、更优选为50000~500000、进一步优选为100000~300000。通过使sis的重均分子量为上述范围,浸渍成型体等膜成型体的拉伸强度与柔软性之间的平衡性提高,并且sis的胶乳有变得容易制造的倾向。
93.sis胶乳中的胶乳颗粒(sis颗粒)的体积平均粒径优选为0.1~10μm、更优选为0.5~3μm、进一步优选为0.5~2.0μm。通过使胶乳颗粒的体积平均粒径为上述范围,胶乳粘度变得适度、变得容易处理,并且能够在贮藏sis胶乳时,抑制胶乳表面生成皮膜。
94.此外,作为共轭二烯系聚合物,也能够使用除去蛋白质的天然橡胶。作为除去蛋白质的天然橡胶的原料的天然橡胶,能够使用从天然橡胶的树中得到的胶乳所包含的天然橡胶、和处理该胶乳后的胶乳所包含的天然橡胶,能够使用例如从天然橡胶的树采集的鲜胶乳(field latex)所包含的天然橡胶、将鲜胶乳通过氨等处理而成的市售的天然橡胶胶乳所包含的天然橡胶等。
95.在除去蛋白质得到天然橡胶时,作为从天然橡胶中除去蛋白质的方法没有特别限定,能够在表面活性剂的存在下,使天然橡胶胶乳与尿素化合物反应,使天然橡胶中包含的蛋白质改性后,对这样的含有改性蛋白质的天然橡胶胶乳实施离心分离、橡胶成分的凝固、超滤等处理来将天然橡胶与改性蛋白质分离,除去该改性蛋白质,由此得到除去蛋白质的天然橡胶的胶乳。
96.此外,作为共轭二烯系聚合物,如上所述,能够使用含腈基共轭二烯系共聚物。
97.含腈基共轭二烯系共聚物是将共轭二烯单体与烯属不饱和腈单体共聚而成的共聚物,除这些之外,也可以是将根据需要而使用的能够与这些共聚的其他的烯属不饱和单体共聚而成的共聚物。
98.作为共轭二烯单体,可举出例如1,3
‑
丁二烯、异戊二烯、2,3
‑
二甲基
‑
1,3
‑
丁二烯、2
‑
乙基
‑
1,3
‑
丁二烯、1,3
‑
戊二烯和氯丁二烯等。在这些中,优选1,3
‑
丁二烯和异戊二烯,更优选1,3丁二烯。这些共轭二烯单体能够单独使用,或组合两种以上使用。在含腈基共轭二烯系共聚物中,通过共轭二烯单体形成的共轭二烯单体单元的含有比例优选为56~78重量%,更优选为56~73重量%,进一步为优选56~68重量%。通过使共轭二烯单体单元的含量为上述范围,能够使得到的浸渍成型体等膜成型体的拉伸强度充分,并且使手感和拉伸率更优异。
99.作为烯属不饱和腈单体,只要是含有腈基的烯属不饱和单体则没有特别限定,可举出例如:丙烯腈、甲基丙烯腈、富马腈、α
‑
氯丙烯腈、α
‑
氰基乙基丙烯腈等。在这些中,优选丙烯腈和甲基丙烯腈,更优选丙烯腈。这些烯属性不饱和腈单体能够单独使用,或组合两种以上使用。在含腈基共轭二烯系共聚物中,通过烯属不饱和腈单体形成的烯属不饱和腈单体单元的含有比例优选为20~40重量%、更优选为25~40重量%、进一步优选为30~40重量%。通过使烯属不饱和腈单体单元的含量为上述范围,能够使得到的浸渍成型体等膜成型体的拉伸强度充分,并且使手感和拉伸率更优异。
100.作为能够与共轭二烯单体和烯属不饱和腈单体共聚的其他的烯属不饱和单体,能够举出例如作为含有羧基的烯属不饱和单体的烯属不饱和羧酸单体;苯乙烯、烷基苯乙烯、乙烯基萘等乙烯基芳香族单体;氟乙基乙烯基醚等氟烷基乙烯基醚;(甲基)丙烯酰胺、n
‑
羟甲基(甲基)丙烯酰胺、n,n
‑
二羟甲基(甲基)丙烯酰胺、n
‑
甲氧基甲基(甲基)丙烯酰胺、n
‑
丙氧基甲基(甲基)丙烯酰胺等烯属不饱和酰胺单体;(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸
‑2‑
乙基己酯、(甲基)丙烯酸三氟乙酯、(甲基)丙烯酸四氟丙酯、马来酸二丁酯、富马酸二丁酯、马来酸二乙酯、(甲基)丙烯酸甲氧基甲酯、(甲基)丙烯酸乙氧基乙酯、(甲基)丙烯酸甲氧基乙氧基乙酯、(甲基)丙烯酸氰基甲酯、(甲基)丙烯酸
‑2‑
氰基乙酯、(甲基)丙烯酸
‑1‑
氰基丙酯、(甲基)丙烯酸
‑2‑
乙基
‑6‑
氰基己酯、(甲基)丙烯酸
‑3‑
氰基丙酯、(甲基)丙烯酸羟基乙酯、(甲基)丙烯酸羟基丙酯、(甲基)丙烯酸缩水甘油酯、(甲基)丙烯酸二甲基氨基乙酯等烯属不饱和羧酸酯单体;二乙烯基苯、聚乙二醇二(甲基)丙烯酸酯、聚丙二醇二(甲基)丙烯酸酯、三羟甲基丙烷三(甲基)丙烯酸酯、季戊四醇(甲基)丙烯酸酯等交联性单体等。这些烯属不饱和单体能够单独使用,或组合两种以上使用。另外,作为能够共聚的其他烯属不饱和单体,能够通过使用烯属不饱和羧酸单体,使含腈基共轭二烯系共聚物具有羧基。
101.作为烯属不饱和羧酸单体,只要是含有羧基的烯属不饱和单体则没有特别限定,
可举出例如丙烯酸、甲基丙烯酸等烯属不饱和单羧酸单体;衣康酸、马来酸、富马酸等烯属不饱和多元羧酸单体;马来酸酐、柠康酸酐等烯属不饱和多元羧酸酐;富马酸单丁酯、马来酸单丁酯、马来酸单
‑2‑
羟基丙酯等烯属不饱和多元羧酸的偏酯单体等。在这些中,优选烯属不饱和单羧酸、特别优选甲基丙烯酸。这些烯属不饱和羧酸单体能够作为碱金属盐或铵盐而使用。此外,这些烯属性不饱和羧酸单体能够单独使用,或组合两种以上使用。在含腈基共轭二烯系共聚物中,通过烯属不饱和羧酸单体形成的烯属不饱和羧酸单体单元的含有比例优选为2~5重量%、更优选为2~4.5重量%、进一步优选为2.5~4.5重量%。通过使烯属不饱和羧酸单体单元的含量为上述范围,能够使得到的浸渍成型体等膜成型体的拉伸强度充分,并且使手感和拉伸率更优异。
102.在含腈基共轭二烯系共聚物中,通过其他的烯属不饱和单体形成的其他的单体单元的含有比例优选为10重量%以下、更优选为5重量%以下、进一步优选为3重量%以下。
103.含腈基共轭二烯系共聚物通过将含有上述的单体形成的单体混合物共聚而得到,优选通过乳液聚合进行共聚的方法。作为乳液聚合的方法,能够采用现有公知的方法。
104.含腈共轭二烯系共聚物的胶乳的数均粒径优选为60~300nm,更优选为80~150nm。粒径能够通过调节乳化剂和聚合引发剂的使用量等方法,调节至所期望的值。
105.此外,作为本发明使用的共轭二烯系聚合物,能够如上所述使用合成聚异戊二烯、苯乙烯
‑
异戊二烯
‑
苯乙烯嵌段共聚物(sis)、除去蛋白质的天然橡胶、含腈基共轭二烯系共聚物等,但不限于这些,可以使用丁二烯聚合物、苯乙烯
‑
丁二烯共聚物等。
106.丁二烯聚合物可以是作为共轭二烯单体的1,3
‑
丁二烯的均聚物,也可以是与能够同作为共轭二烯单体的1,3
‑
丁二烯共聚的其他烯属不饱和单体共聚而形成的共聚物。
107.此外,苯乙烯
‑
丁二烯共聚物是将作为共轭二烯单体的1,3
‑
丁二烯与苯乙烯共聚而成的共聚物,除这些之外,也可以是将根据需要而使用的能够与这些共聚的其他的烯属不饱和单体共聚形成的共聚物。
108.此外,本发明使用的共轭二烯系聚合物可以是经羧基改性的羧基改性共轭二烯系聚合物。羧基改性共轭二烯系聚合物能够通过利用具有羧基的单体将上述的共轭二烯系聚合物进行改性来得到。另外,作为含腈基共轭二烯系共聚物,在使用烯属不饱和羧酸单体作为可行的其他烯属不饱和单体的情况下,因为已经进行了羧基改性,因此不一定需要利用后述具有羧基的单体进行改性。
109.作为利用具有羧基的单体将共轭二烯系聚合物进行改性的方法,没有特别限定,可举出例如将共轭二烯系聚合物与具有羧基的单体在水相中进行接枝聚合的方法。作为将共轭二烯系聚合物与具有羧基的单体在水相中进行接枝聚合的方法没有特别限定,使用现有公知的方法即可,优选例如在共轭二烯系聚合物的胶乳中添加具有羧基的单体和有机过氧化物后,在水相中使共轭二烯系聚合物与具有羧基的单体反应的方法。
110.作为有机过氧化物没有特别限定,可举出例如二异丙基苯过氧化氢、异丙基苯过氧化氢、叔丁基过氧化氢、1,1,3,3
‑
四甲基丁基过氧化氢、二叔丁基过氧化物、异丁酰基过氧化物、苯甲酰基过氧化物等,从提高得到的浸渍成型体的机械强度的观点出发,特别优选1,1,3,3
‑
四甲基丁基过氧化氢。这些有机过氧化物可以单独使用一种,也可以组合使用两种以上。
111.有机过氧化物的添加量没有特别限定,相对于100重量份的共轭二烯系聚合物的
胶乳所包含的共轭二烯系聚合物,优选为0.01~3重量份、更优选为0.1~1重量份。
112.此外,有机过氧化物能够与还原剂组合,作为氧化还原系聚合引发剂使用。作为还原剂没有特别限定,可举出硫酸亚铁、环烷酸亚铜等含有处于还原状态的金属离子的化合物;羟甲烷亚磺酸钠等亚磺酸盐;二甲基苯胺等胺化合物等。这些还原剂可以单独使用一种,也可以组合使用两种以上。
113.还原剂的添加量没有特别限定,相对于1重量份的有机过氧化物,优选为0.01~1重量份。
114.有机过氧化物和还原剂的添加方法没有特别限定,能够分别使用一次性添加、分次添加、连续添加等公知的添加方法。
115.作为使共轭二烯系聚合物与具有羧基的单体进行反应时的反应温度没有特别限定,优选为15~80℃,更优选为30~50℃。使共轭二烯系聚合物与具有羧基的单体反应时的反应时间根据上述反应温度适当设定即可,优选为30~300分钟、更优选为60~120分钟。
116.作为使共轭二烯系聚合物与具有羧基的单体反应时的共轭二烯系聚合物的胶乳的固体成分浓度,没有特别限定,优选为5~60重量%、更优选为10~40重量%。
117.作为具有羧基的单体,可举出例如丙烯酸、甲基丙烯酸等烯属不饱和单羧酸单体;衣康酸、马来酸、富马酸、丁烯三羧酸等烯属不饱和多元羧酸单体;富马酸单丁酯、马来酸单丁酯、马来酸单2
‑
羟基丙酯等烯属不饱和多元羧酸的偏酯;马来酸酐、柠檬酸酐等多元羧酸酐等,从使羧基改性的效果更进一步显著的方面出发,优选烯属不饱和单羧酸单体,特别优选丙烯酸和甲基丙烯酸。另外,这些单体可以单独使用一种,也可以并用两种以上。此外,上述羧基也包含与碱金属、铵等形成盐的羧基。
118.具有羧基的单体的使用量相对于100质量份的共轭二烯系聚合物,优选为0.01重量份~100重量份、更优选为0.01重量份~40重量份、进一步优选为0.5重量份~20重量份。通过使具有羧基的单体的使用量为上述范围,得到的共轭二烯系聚合物胶乳组合物的粘度更适度,容易运送,且得到的浸渍成型体等膜成型体的拉伸强度进一步提高。
119.作为将具有羧基的单体添加至共轭二烯系聚合物的胶乳中的方法,没有特别限定,能够采用一次性添加、分次添加、连续添加等公知的添加方法。
120.羧基改性共轭二烯系聚合物中具有羧基的单体的改性率根据得到的共轭二烯系聚合物胶乳组合物的使用目的适当控制即可,优选为0.01~10重量%、更优选为0.2~5重量%、进一步优选为0.3~3重量%、特别优选为0.4~2重量%。另外,改性率通过下述式表示。
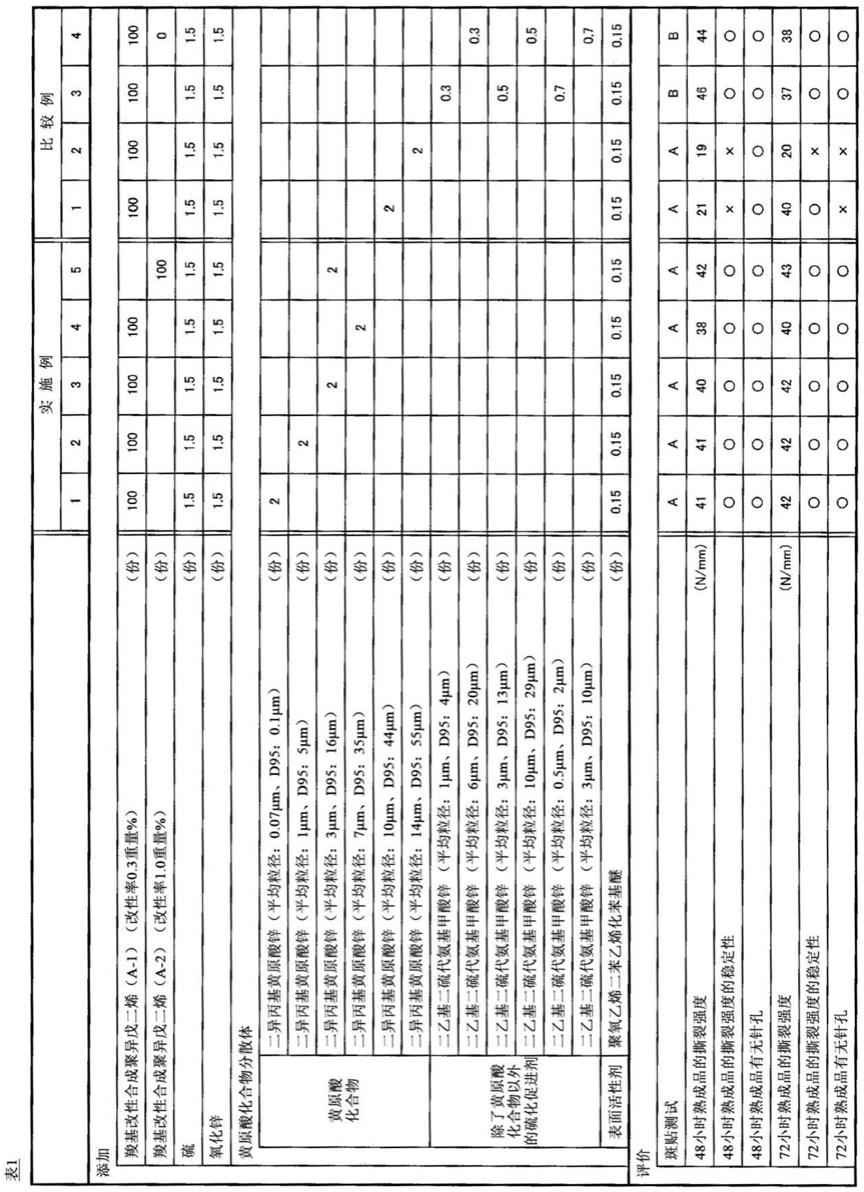
121.改性率(重量%)=(x/y)
×
100
122.另外,在上述式中,x表示羧基改性共轭二烯系聚合物中具有羧基的单体单元的重量,y表示羧基改性共轭二烯系聚合物的重量。x能够通过如下的方法求出:对羧基改性共轭二烯系聚合物进行1h
‑
nmr测定,根据1h
‑
nmr测定的结果算出,或通过中和滴定求出酸量,根据酸量算出。
123.作为用于接枝聚合的聚合催化剂(接枝聚合催化剂)没有特别限定,能够举出例如过硫酸钠、过硫酸钾、过硫酸铵、过磷酸钾、过氧化氢等无机过氧化物;二异丙基苯过氧化氢、异丙基苯过氧化氢、叔丁基过氧化氢、1,1,3,3
‑
四甲基丁基过氧化氢、二叔丁基过氧化物、异丁酰基过氧化物、苯甲酰过氧化物等有机过氧化物;2,2
’‑
偶氮二异丁腈、偶氮双
‑
2,
4
‑
二甲基戊腈、偶氮二异丁酸甲酯等偶氮化合物等,从得到的浸渍成型体等膜成型体的拉伸强度进一步提高的观点出发,优选有机过氧化物,特别优选1,1,3,3
‑
四甲基丁基过氧化氢。
124.上述接枝聚合催化剂能够分别单独使用,或组合使用两种以上。接枝聚合催化剂的使用量因其种类而不同,相对于100重量份的共轭二烯系聚合物,优选为0.1~10重量份、更优选为0.2~5重量份。作为添加接枝聚合催化剂的方法没有特别限定,能够采用一次性添加、分次添加、连续添加等公知的添加方法。
125.可以在本发明使用的共轭二烯系聚合物(包含羧基改性的共轭二烯系聚合物)的胶乳中添加胶乳领域通常添加的ph调节剂、消泡剂、防腐剂、螯合剂、除氧剂、分散剂、抗老化剂等添加剂。
126.作为ph调节剂,可举出例如氢氧化钠、氢氧化钾等碱金属的氢氧化物;碳酸钠、碳酸钾等碱金属的碳酸盐;碳酸氢钠等碱金属的碳酸氢盐;氨;三甲胺、三乙醇胺等有机胺化合物等,优选碱金属氢氧化物或氨。
127.本发明使用的共轭二烯系聚合物(包含羧基改性的共轭二烯系聚合物)的胶乳的固体成分浓度优选为30~70重量%、更优选为40~70重量%。通过使固体成分浓度为上述范围,能够更有效地抑制胶乳中的凝聚物的产生,且能够在贮藏胶乳时更有效地抑制聚合物颗粒的分离。
128.本发明的共轭二烯系聚合物胶乳组合物除上述的共轭二烯系聚合物的胶乳以外,还含有上述的本发明的黄原酸化合物分散体和硫化剂。
129.本发明的共轭二烯系聚合物胶乳组合物中的黄原酸化合物分散体的添加量没有特别限定,相对于100重量份的共轭二烯系聚合物,黄原酸化合物的含量优选为0.01~10重量份的量、更优选为0.1~7重量份的量、进一步优选为为0.5~5重量份的量。通过使黄原酸化合物分散体的添加量为上述范围,对于得到的浸渍成型体等膜成型体,能够抑制迟发型过敏(type iv)症状的产生,并使撕裂强度进一步提高。
130.另外,黄原酸化合物分散体中所包含的黄原酸化合物通过本发明的共轭二烯系聚合物胶乳组合物所包含的活化剂的作用,以黄原酸盐的形式存在,结果在共轭二烯系聚合物胶乳组合物中可以包含两种以上的黄原酸化合物。或者在共轭二烯系聚合物胶乳组合物中包含硫作为硫系硫化剂等的情况下,在共轭二烯系聚合物胶乳组合物中,黄原酸化合物通过硫的作用,其一部分可以以黄原酸二硫化物、黄原酸多硫化物的形式存在。
131.作为硫化剂,优选使用硫系硫化剂。作为硫系硫化剂,可举出例如硫粉、升华硫、沉淀硫、胶体硫、表面处理硫、不溶性硫等硫;氯化硫、二氯化硫、二硫化吗啉、二硫化烷基酚、二硫化己内酰胺(n,n'
‑
二硫代双(六氢
‑
2h
‑
氮杂
‑
2))、含磷多硫化物、高分子多硫化物、2
‑
(4
’‑
吗啉代二硫代)苯并噻唑等含硫化合物。在这些中,能够优选使用硫。硫系加硫剂能够单独使用一种,或组合使用两种以上。
132.本发明的胶乳组合物中的硫化剂的含量没有特别限定,相对于100重量份的共轭二烯系聚合物,优选为0.1~10重量份、更优选为0.2~5重量份、进一步优选为0.5~3重量份。通过使硫化剂的含量为上述范围,在得到的浸渍成型体等的膜成型体中,能够抑制硬度的提高,并且更加提高拉伸强度。
133.此外,本发明的共轭二烯系聚合物胶乳组合物优选还含有活化剂。
134.作为活化剂,只要是通过与黄原酸化合物分散体所包含的黄原酸化合物一起使用来作为活化剂发挥作用的化合物即可,没有特别限定,可举出例如金属氧化物。在使用羧基改性共轭二烯系聚合物作为共轭二烯系聚合物的情况下,金属氧化物也作为将羧基交联的交联剂发挥作用,由此能够进一步提高得到的浸渍成型体等膜成型体的拉伸强度,因而优选。
135.作为金属氧化物没有特别限定,可举出氧化锌、氧化镁、氧化钛、氧化钙、铅氧化物、氧化铁、氧化铜、氧化锡、氧化镍、氧化铬、氧化钴和氧化铝等。在这些中,从得到的浸渍成型体等膜成型体的拉伸强度进一步提高的观点出发,优选氧化锌。这些金属氧化物可以单独使用一种,也可以并用多种。
136.本发明的共轭二烯系聚合物胶乳组合物中活化剂的含量没有特别限定,相对于100重量份的共轭二烯系聚合物,优选为0.01~30重量份、更优选为0.1~10重量份、进一步优选为0.5~5重量份。通过使活化剂的含量为上述范围,能够使得到的浸渍成型体等膜成型体的拉伸强度进一步提高。
137.此外,对于本发明的共轭二烯系聚合物胶乳组合物,如果在能够抑制得到的浸渍成型体等膜成型体中的迟发型过敏(type iv)症状的产生的范围,可以还含有硫化促进剂。
138.作为硫化促进剂,能够使用在浸渍成型等膜成型中通常所使用的硫化促进剂,可举出例如:二乙基二硫代氨基甲酸、二丁基二硫代氨基甲酸、二
‑2‑
乙基己基二硫代氨基甲酸、二环己基二硫代氨基甲酸、二苯基二硫代氨基甲酸、二苄基二硫代氨基甲酸等二硫代氨基甲酸类及它们的锌盐;2
‑
巯基苯并噻唑、2
‑
巯基苯并噻唑锌、2
‑
巯基噻唑啉、二苯并噻唑基二硫醚、2
‑
(2,4
‑
二硝基苯硫基)苯并噻唑、2
‑
(n,n
‑
二乙基硫代氨基甲酰基硫代)苯并噻唑、2
‑
(2,6
‑
二甲基
‑4‑
吗啉基硫代)苯并噻唑、2
‑
(4
’‑
吗啉基二硫代)苯并噻唑、4
‑
吗啉基
‑2‑
苯并噻唑基二硫醚、1,3
‑
双(2
‑
苯并噻唑基巯基甲基)脲等。但是,从适当地抑制迟发型过敏(type iv)症状的产生的观点出发,优选使用除秋兰姆系硫化促进剂、二硫代氨基甲酸盐系硫化促进剂、噻唑系硫化促进剂以外的硫化促进剂。另外,硫化促进剂能够单独使用一种,或组合使用两种以上。
139.本发明的共轭二烯系聚合物胶乳组成物能够根据需要进一步添加抗老化剂;分散剂;炭黑、二氧化硅、滑石等增强剂;碳酸钙、黏土等填充剂;紫外线吸收剂;增塑剂等添加剂。
140.作为抗老化剂,可举出2,6
‑
二
‑4‑
甲基苯酚、2,6
‑
二叔丁基苯酚、丁基羟基苯胺、2,6
‑
二叔丁基
‑
α
‑
二甲基胺基对甲酚、十八烷基
‑3‑
(3,5
‑
二叔丁基
‑4‑
羟基苯基)丙酸酯、苯乙烯化苯酚、2,2
’‑
亚甲基
‑
双(6
‑
α
‑
甲基苄基对甲酚)、4,4
’‑
亚甲基双(2,6
‑
二叔丁基苯酚)、2,2
’‑
亚甲基
‑
双(4
‑
甲基
‑6‑
叔丁基苯酚)、烷基化双酚、对甲酚和二环戊二烯的丁基化反应生成物等不含有硫原子的酚系抗老化剂;2,2
’‑
硫代双
‑
(4
‑
甲基
‑6‑
叔丁基苯酚)、4,4
’‑
硫代双
‑
(6
‑
叔丁基邻甲酚)、2,6
‑
二叔丁基
‑4‑
(4,6
‑
双(辛基硫代)
‑
1,3,5
‑
三嗪
‑2‑
基氨基)苯酚等硫代双酚系抗老化剂;三(壬基苯基)亚磷酸酯、二苯基异癸基亚磷酸酯、四苯基二丙二醇二亚磷酸酯等亚磷酸酯系抗老化剂;硫代二丙酸二月桂酯等硫酯系抗老化剂;苯基
‑
α
‑
萘胺、苯基
‑
β
‑
萘胺、对(对苯甲磺酰胺)二苯胺、4,4
’‑
(α,α
‑
二甲基苄基)二苯胺、n,n
‑
二苯基对亚苯基二胺、n
‑
异丙基
‑
n
’‑
苯基对亚苯基二胺、丁醛苯胺缩合物等胺系抗老化剂;6
‑
乙氧基
‑
2,2,4
‑
三甲基
‑
1,2
‑
二氢喹啉等喹啉系抗老化剂;2,5
‑
二(叔戊基)氢醌等氢醌系抗老化
剂等。这些抗老化剂能够单独使用一种,或并用两种以上。
141.本发明的共轭二烯系聚合物胶乳组合物中的抗老化剂的含量相对于100重量份的羧基改性共轭二烯系聚合物,优选为0.05~10重量份、更优选为0.1~5重量份。
142.作为制备本发明的共轭二烯系聚合物胶乳组合物的方法,没有特别限定,可举出例如在上述的共轭二烯系聚合物的胶乳中混合活化剂和黄原酸化合物分散体、以及根据需要使用的各种添加剂的方法等。此时,也能够采用在制备除共轭二烯系聚合物的胶乳以外的添加成分的水性分散液后,将该水性分散液与共轭二烯系聚合物的胶乳进行混合的方法等。
143.另外,本发明的共轭二烯系聚合物胶乳组合物的固体成分浓度优选为10~60重量%、更优选为10~55重量%。
144.此外,在本发明中,从使得到的浸渍成型体等膜成型体的机械特性充分的观点出发,优选在供给至浸渍成型等膜成型前,对本发明的共轭二烯系聚合物胶乳组合物进行熟化(前硫化)。熟化(前硫化)的时间没有特别限定,优选为6~70小时,更优选为6~60小时,进一步优选为6~50小时。另外,在制备共轭二烯系聚合物胶乳组合物时,在使用还含有非离子系表面活性剂和/或非离子阴离子系表面活性剂的分散体作为黄原酸化合物分散体的情况下,即使将熟化(前硫化)时间缩短至优选6~30小时、更优选6~18小时的情况下,也能够得到具有充分的机械特性的浸渍成型体等膜成型体,由此能够缩短实现熟化(前硫化)所需要的时间和进一步提高生产效率。另外,前硫化的温度没有特别限定,优选为20~40℃。
145.<膜成型体>
146.本发明的膜成型体是由上述的本发明的共轭二烯系聚合物胶乳组合物形成的膜状的成型体。本发明的膜成型体的膜厚优选为0.03~0.50mm,更优选为0.05~0.40mm,特别优选为0.08~0.30mm。
147.作为本发明的膜成型体没有特别限定,优选为将本发明的共轭二烯系聚合物胶乳组合物浸渍成型而得到的浸渍成型体。浸渍成型采用如下的方法:将模具浸渍至共轭二烯系聚合物胶乳组合物,使该组合物在模具的表面沉积,接下来将模具从该组合物中提起,然后使沉积在模具表面的该组合物干燥。另外,可以对浸渍至共轭二烯系聚合物胶乳组合物之前的模具进行预热。此外,在将模具浸渍至共轭二烯系聚合物胶乳组合物之前,或者在将模具从共轭二烯系聚合物胶乳组合物提起后,能够根据需要而使用凝固剂。
148.作为凝固剂的使用方法的具体例子,有将浸渍至共轭二烯系聚合物胶乳组合物之前的模具浸渍至凝固剂的溶液中使凝固剂附着的方法(阳极凝结浸渍法)、将沉积了共轭二烯系聚合物胶乳组合物的模具浸渍至凝固剂溶液中的方法(迪克(teague)凝结浸渍法)等,从得到厚度不均少的浸渍成型体的方面出发,优选阳极凝结浸渍法。
149.作为凝固剂的具体例子,有氯化钡、氯化钙、氯化镁、氯化锌、氯化铝等卤化金属;硝酸钡、硝酸钙、硝酸锌等硝酸盐;乙酸钡、乙酸钙、乙酸锌等乙酸盐;硫酸钙、硫酸镁、硫酸铝等硫酸盐等水溶性多价金属盐。其中优选钙盐,更优选硝酸钙。这些水溶性多元金属盐能够单独使用一种,或并用两种以上。
150.凝固剂通常能够使用水、酒精或这些的混合物的溶液,优选以水溶液的状态使用。该水溶液可以进一步含有甲醇、乙醇等水溶性有机溶剂、非离子性表面活性剂。凝固剂的浓度根据水溶性多价金属盐的种类而不同,优选为5~50重量%、更优选为10~30重量。
151.在将模具从共轭二烯系聚合物胶乳组合物中提起后,通常进行加热以使形成在模具上的沉积物干燥。干燥条件适当选择即可。
152.接下来,对得到的浸渍成型层实施加热处理,进行硫化。此外,此时在实施加热处理前,可以在水中、优选为30~70℃的温水中浸渍1~60分钟左右,除去水溶性杂质(例如,剩余的乳化剂、凝固剂等)。水溶性杂质的除去操作可以在对浸渍成型层进行加热处理后进行,从能够有效地除去水溶性杂质的方面出发,优选在加热处理前进行。
153.浸渍成型层的硫化通常在80~150℃的温度下优选通过实施10~130分钟的加热处理来进行。作为加热方法,能够采用通过红外线、加热空气进行的外部加热或通过高频率进行的内部加热的方法。其中,优选通过加热空气进行的外部加热。
154.而且,通过将浸渍成型层从浸渍成型用模具脱离,可以得到作为膜状的膜成型体的浸渍成型体。作为脱离方法,能够采用用手从成型用模具剥离,或通过水压、压缩空气进行剥离的方法。另外,在脱离后,也可以进一步在60~120℃的温度进行10~120分的加热处理。
155.另外,本发明的膜成型体除上述的将本发明的胶乳组合物浸渍成型的方法之外,只要是能够将上述的本发明的胶乳组合物成型为膜状的方法(例如,涂布法等),可以通过任意方法得到。
156.包含本发明的浸渍成型体的本发明的膜成型体由于是使用上述的本发明的共轭二烯系聚合物胶乳组合物而得到的膜成型体,因此熟化(前硫化)所需要的时间缩短,生产率优异,而且撕裂强度也优异,能够特别优选作为手套使用。在膜成型体为手套的情况下,为了防止膜成型体彼此的接触面的密合,改善穿脱时的滑动,可以将滑石、碳酸钙等无机微粒或淀粉颗粒等有机微颗粒撒布于手套表面,或在手套表面形成含有微粒的弹性体层,或将手套的表面层氯化。
157.此外,含有本发明的浸渍成型体的本发明的膜成型体除上述手套之外,还能够用于奶瓶用奶嘴、滴液吸管、管子、水枕、气球气囊、医用导管、安全套等医疗用品;气球、人偶、球等玩具;加压成型用袋、气体贮藏用袋等工业用品;指套等。
158.<粘接剂组合物>
159.此外,在本发明中,能够将上述的本发明的共轭二烯系聚合物胶乳组合物用作粘接剂组合物。
160.粘接剂组合物中的本发明的共轭二烯系聚合物胶乳组合物的含量(固体成分量)优选为5~60重量%,更优选为10~30重量%。
161.粘接剂组合物除本发明的共轭二烯系聚合物胶乳组合物以外,优选还含有粘接剂树脂。作为粘接剂树脂没有特别限定,能够优选使用例如间苯二酚
‑
甲醛树脂、三聚氰胺树脂、环氧树脂和异氰酸酯树脂,在这些中,优选间苯二酚
‑
甲醛树脂。间苯二酚
‑
甲醛树脂能够使用公知的间苯二酚
‑
甲醛树脂(例如日本特开昭55
‑
142635号公报公开的间苯二酚
‑
甲醛树脂)。间苯二酚与甲醛的反应比率以“间苯二酚∶甲醛”的摩尔比计,通常为1∶1~1∶5,优选为1∶1~1∶3。
162.此外,为了进一步提高粘接剂组合物的粘接力,能够使粘接剂组合物含有一直以来使用的2,6
‑
双(2,4
‑
二羟基苯基甲基)
‑4‑
氯苯酚或类似的化合物、异氰酸酯、封闭异氰酸酯、乙烯脲、聚环氧化物、改性聚氯乙烯树脂等。
163.进而,能够使粘接剂组合物含有交联助剂。通过使其含有交联助剂,能够令使用粘接剂组合物得到的后述复合体的机械强度提高。作为交联助剂,能够举出对醌二肟等醌二肟;甲基丙烯酸月桂酯、甲基丙烯酸甲酯等甲基丙烯酸酯;daf(富马酸二烯丙酯)、dap(邻苯二甲酸二烯丙酯)、tac(氰尿酸三烯丙酯)、taic(异氰尿酸三烯丙酯)等烯丙基化合物;双马来酰亚胺、苯基马来酰亚胺、n,n
‑
间亚苯基二马来酰亚胺等马来酰亚胺化合物;硫等。
164.<粘接剂层形成基材>
165.本发明的粘接剂层形成基材通过将使用上述的本发明的共轭二烯系聚合物胶乳组合物或粘接剂组合物形成的粘接剂层形成在基材表面来得到。
166.作为基材没有特别限定,能够使用例如纤维基材。构成纤维基材的纤维的种类没有特别限定,可举出例如维纶纤维、聚酯纤维、尼龙纤维、芳纶(芳香族聚酰胺)等聚酰胺纤维、玻璃纤维、棉、人造丝等。这些能够根据其用途而进行适当选择。纤维基材的形状没有特别限定,能够举出例如短纤维状、丝状、线状、绳状、织布(帆布等)等,能够根据其用途而进行适当选择。例如,粘接剂层形成基材能够经由粘接剂层与橡胶粘接,由此作为基材
‑
橡胶复合体而使用。作为基材
‑
橡胶复合体,没有特别限定,可举出例如使用线状的纤维基材作为纤维基材的带芯线的橡胶制同步带、使用帆布等底布状的纤维基材的橡胶制同步带等。
167.作为得到基材
‑
橡胶复合体的方法,没有特别限定,可举出例如通过浸渍处理等使基材附着共轭二烯系聚合物胶乳组合物或粘接剂组合物而得到粘接剂层形成基材、将粘接剂层形成基材载置在橡胶上,对其进行加热和加压的方法。加压能够使用压缩(压制)成型机、金属辊、注射成型机等进行。此外,加压的压力优选为0.5~20mpa、更优选为2~10mpa。此外,加热的温度优选130~300℃、更优选150~250℃。加热和加压的处理时间优选为1~180分钟、更优选为5~120分钟。通过加热和加压的方法,能够同时进行橡胶的成型、以及粘接剂层形成基材与橡胶的粘接。另外,在加压使用的压缩机的模具的内面、辊的表面优选预先形成用于对作为目标的基材
‑
橡胶复合体的橡胶赋予期望的表面形状的模具。
168.此外,作为基材
‑
橡胶复合体的一个方式,能够举出基材
‑
橡胶
‑
基材复合体。基材
‑
橡胶
‑
基材复合体能够通过例如将基材(可以是两种以上的基材的复合体)与基材
‑
橡胶复合体进行组合而形成。具体而言,将作为基材的芯线、橡胶和作为基材的底布重叠(此时,预先使芯线和底布适当附着共轭二烯系聚合物胶乳组合物或粘接剂组合物,制成粘接剂层形成基材),一边加热一边加压,由此能够得到基材
‑
橡胶
‑
基材复合体。
169.使用本发明的粘接剂层形成基材得到的基材
‑
橡胶复合体是机械强度、耐磨损性和耐水性优异的复合体,因此能够优选作为平形带、v形带、v形多楔带、圆形带、方形带、同步带等带而使用。此外,使用本发明的粘接剂层形成基材得到的基材
‑
橡胶复合体的耐油性优异,能够优选地用作油中带。进而,使用本发明的粘接剂层形成基材而得到的基材
‑
橡胶复合体也能够优选地用于软管、管、膜片等。作为软管,可举出单管橡胶软管、多层橡胶软管、编制式增强软管、夹布式增强软管等。作为膜片(diaphragm),可举出平膜片、滚动膜片等。
170.使用本发明的粘接剂层形成基材得到的基材
‑
橡胶复合体除上述的用途以外,还能够作为密封件、橡胶辊等工业用制品使用。作为密封件,可举出旋转用、摇动用、往复运动等运动部位密封件和固定部位密封件。作为运动部位密封件,可举出油密封件、活塞密封件、机械密封件、保护罩、防尘罩、膜片、蓄压器等。作为固定部位密封件,可举出o型环、各种
垫片等。作为橡胶辊,可举出印刷机器、复印机器等办公自动化机器的部件的辊;纺丝用拉伸辊、纺织用牵伸辊等纤维加工用辊;张力辊、缓冲辊、转向辊等制铁用辊等。
171.实施例
172.以下,通过实施例对本发明进行详细地说明,但是本发明并不限于这些实施例。另外,只要没有特别说明,以下的“份”为重量基准。另外,各种物性如下进行测定。
173.<固体成分浓度>
174.在铝盘(重量:x1)中精确称量2g的试样(重量:x2),将其在105℃的热风干燥器内干燥2小时。接下来,在干燥器内冷却后,测定铝盘整体的重量(重量:x3),根据下式算出固体成分浓度。
175.固体成分浓度(重量%)=(x3
‑
x1)
×
100/x2
176.<羧基改性合成聚异戊二烯的改性率>
177.使用氢氧化钠水溶液对构成羧基改性合成聚异戊二烯的胶乳的羧基改性合成聚异戊二烯进行中和滴定,由此求出羧基改性合成聚异戊二烯中的羧基数。接下来,基于求出的羧基数,按照下述式求出具有羧基的单体的改性率。
178.改性率(重量%)=(x/y)
×
100
179.另外,在上述式中,x表示羧基改性合成聚异戊二烯中具有羧基的单体单元的重量,y表示羧基改性合成聚异戊二烯的重量。
180.<斑贴测试>
181.将膜厚约0.2mm的膜状的浸渍成型体切断成10
×
10mm的大小,得到试验片,将该试验片分别粘贴在10名受试者的手臂上。然后,在48小时后观察粘贴部分,由此确认有无迟发型过敏(type iv)的过敏症状产生,按照以下的基准进行评价。
182.另外,斑贴测试使用熟化(预硫化)时间为48小时的浸渍成型体进行。
183.a:所有受试者均未发现过敏症状。
184.b:一部分受试者发现有过敏症状。
185.<浸渍成型体的撕裂强度、撕裂强度的稳定性>
186.根据astm d624
‑
00,将浸渍成型体在23℃、相对湿度50%的恒温恒湿室放置24小时以上后,用哑铃型裁刀(商品名“die c”,dumbbell公司制)冲裁,制作撕裂强度测定用的测试片。然后,用tensilon万能试验机(商品名“rtc
‑
1210”、a&d公司制)将该试验片以500mm/min的拉伸速度拉伸,测定撕裂强度(单位:n/mm)。另外,测定对5个试验片进行,在5个试验片的撕裂强度的测定值中,将中位数(即,5个试验片中显示第3大的值的试验片的撕裂强度的值)作为撕裂强度的值采用。此外,根据5个试验片的撕裂强度的测定值,按照下述基准评价撕裂强度的稳定性。
187.〇:撕裂强度的测定值落入相对于中位数
±
10%的范围的试验片的比例为70%以上(即,在5个试验片中,撕裂强度的测定值落入相对于中位数
±
10%的范围的试验片的个数为4个以上)
188.×
:撕裂强度的测定值落入相对于中位数
±
10%的范围的试验片的比例小于70%(即,在5个试验片中,撕裂强度的测定值落入相对于中位数
±
10%的范围的试验片的个数为3个以下)
189.另外,浸渍成型体的撕裂强度、撕裂强度的稳定性的测定在实施例1~5、比较例1
t
‑
45”,花王公司制)进行稀释,历经5分钟将该稀释液添加至合成聚异戊二烯的胶乳(e)中。接下来,将添加了分散剂的合成聚异戊二烯的胶乳(e)加入氮置换的带搅拌机的反应器中,一边搅拌一边将温度升温至30℃。此外,使用另外的容器混合2份的作为具有羧基的单体的甲基丙烯酸和16份的蒸馏水,制备甲基丙烯酸稀释液。历经30分钟在加热到30℃的反应容器内添加该甲基丙烯酸稀释液。
203.进而,使用另外的容器,制备由7份蒸馏水、0.32份的甲醛次硫酸钠(商品名“sfs”,mitsubishi gas公司制)、0.01份的硫酸亚铁(商品名“frost fe”,中部chelest公司制)组成的溶液(f)。将该溶液(f)转移至反应容器内后,添加0.5份的1,1,3,3
‑
四甲基丁基过氧化氢(商品名“perocta h”,日本油脂公司制),在30℃反应1小时,由此得到羧基改性合成聚异戊二烯(a
‑
1)的胶乳。接下来,通过离心分离机浓缩羧基改性合成聚异戊二烯(a
‑
1),得到固体成分浓度56%的轻液。然后,对得到的羧基改性合成聚异戊二烯(a
‑
1)的胶乳按照上述方法测定具有羧基的单体的改性率,结果改性率为0.3重量%。
204.<制造例2>
205.(羧基改性合成聚异戊二烯(a
‑
2)的胶乳的制造)
206.将甲基丙烯酸的使用量变更为5份,除此以外,与制造例1同样地进行,得到固体成分浓度为55%的羧基改性合成聚异戊二烯(a
‑
2)的胶乳。对得到的羧基改性合成聚异戊二烯(a
‑
2)的胶乳按照上述方法测定具有羧基的单体的改性率,结果改性率为1.0重量%。
207.<制造例3>
208.(羧基改性合成聚异戊二烯(a
‑
3)的胶乳的制造)
209.将甲基丙烯酸的使用量变更为3份,除此以外,与制造例1同样地进行,得到固体成分浓度为55%的羧基改性合成聚异戊二烯(a
‑
3)的胶乳。对得到的羧基改性合成聚异戊二烯(a
‑
3)的胶乳按照上述方法测定具有羧基的单体的改性率,结果改性率为0.5重量%。
210.<实施例1>
211.(黄原酸化合物分散体的制备)
212.通过珠磨机(商品名“star mill lmz
‑
015”,ashizawa finetech ltd.制)混合2份的作为黄原酸化合物的二异丙基黄原酸锌(商品名“nocceler zix”,大内新兴化学工业株式会社制,体积平均粒径:14μm,95%体积累积径(d95):55μm)、0.15份的作为非离子系表面活性剂的聚氧乙烯二苯乙烯化苯基醚(商品名“emulgen a
‑
60”,花王公司制)和4.5份的水,进行破碎处理,由此得到黄原酸化合物分散体。另外,作为利用珠磨机的混合条件,使用的陶瓷制珠、以3800rpm进行1.5小时。此外,使用激光衍射散射式粒度分布计(商品名“sald
‑
2300”,株式会社岛津制作所制)测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为0.07μm、95%体积累积径(d95)为0.1μm。
213.(胶乳组合物的制备)
214.首先,准备苯乙烯
‑
马来酸单仲丁酯
‑
马来酸单甲酯聚合物(商品名“scripset550”,hercules inc.制),使用氢氧化钠中和聚合物中的100%的羧基,由此制备钠盐水溶液(浓度10重量%)。然后,在制造例1中得到的羧基改性合成聚异戊二烯(a
‑
1)的胶乳中,以相对于胶乳中100份的羧基改性合成聚异戊二烯(a
‑
1)以固体成分换算计为0.8份的方式添加该钠盐水溶液,得到混合物。
215.然后,一边搅拌得到的混合物,一边相对于100份的混合物中的羧基改性合成聚异戊二烯(a
‑
1)添加6.65份的上述制备的黄原酸化合物分散体(以二异丙基黄原酸锌换算计为2份)。
216.接下来,以按固体成分换算计为1.5份的作为活化剂的氧化锌、1.5份的硫、2份的抗老化剂(商品名“wingstay l”、goodyear公司制)的方式,添加各添加剂的水分散液,得到胶乳组合物。然后,将得到的胶乳组合物分成2部分,对一部分用调节至25℃的恒温水槽进行48小时熟化(前硫化),对另一部分用调节至25℃的恒温水槽进行72小时熟化(前硫化),由此得到48小时熟化胶乳组合物和72小时熟化胶乳组合物。
217.(浸渍成型体的制造)
218.清洗市售的陶瓷制手形模具(shinko公司制),在70℃的炉内预先加热后,在含有18重量%的硝酸钙和0.05重量%的聚氧乙烯月桂基醚(商品名“emulgen 109p”,花王公司制)的凝固剂水溶液中浸渍5秒,从凝固剂水溶液中取出。接下来,通过将手形模具在70℃的烘箱内干燥30分钟以上,使凝固剂附着在手形模具上,将手形模具用凝固剂覆盖。
219.然后,将覆盖凝固剂的手形模具从烘箱中取出,在上述得到的48小时熟化胶乳组合物中浸渍10秒。接下来,将该手形模具在室温风干10分钟,然后在60℃的温水中浸渍5分钟,使水溶性杂质溶出,在手形模具上形成浸渍成型体。然后,将形成在手形模具上的浸渍成型体通过烘箱以温度130℃、30分钟的条件加热,由此使其硫化后,冷却至室温,撒上滑石后从手形模具剥离,得到手套形状的浸渍成型体(48小时熟化品)。此外,代替48小时熟化胶乳组合物,使用72小时熟化胶乳组合物,除此以外,与上述同样地进行,得到手套形状的浸渍成型体(72小时熟化品)。然后,使用得到的浸渍成型体(48小时熟化品和72小时熟化品),按照上述方法进行斑贴测试、撕裂强度、撕裂强度的稳定性和针孔的有无的各测定、评价。结果示于表1。
220.<实施例2>
221.使用行星式球磨机(商品名“classic line p
‑
5”,fritsch公司制)进行制造黄原酸化合物分散体时的破碎处理,并使用的陶瓷制球、以340rpm进行1小时的条件利用行星式球磨机进行混合,除此以外,与实施例1同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为1μm、95%体积累积径(d95)为5μm。
222.然后,使用得到的黄原酸化合物分散体,除此以外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
223.<实施例3>
224.使用球磨机(商品名“瓷制球磨机”,日陶科学公司制)进行制备黄原酸化合物分散体时的破碎处理,并使用的陶瓷制球(混合了的陶瓷制球(混合了和的陶瓷制球),以50rpm、72小时的条件利用球磨机进行混合,除此以外,与实施例1同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和
95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为3μm、95%体积累积径(d95)为16μm。
225.然后,使用得到的黄原酸化合物分散体,除此以外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
226.<实施例4>
227.将制备黄原酸化合物分散体时利用球磨机的破碎处理的时间变更为48小时,除此以外,与实施例3同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为7μm、95%体积累积径(d95)为35μm。
228.然后,使用得到的黄原酸化合物分散体,除此以外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
229.<实施例5>
230.代替制造例1中得到的羧基改性合成聚异戊二烯(a
‑
1)的胶乳而使用通过制造例2中得到的羧基改性合成聚异戊二烯(a
‑
2)的胶乳,除此之外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
231.<比较例1>
232.将制备黄原酸化合物分散体时利用球磨机的破碎处理的时间变更为24小时,除此以外,与实施例3同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为10μm、95%体积累积径(d95)为44μm。
233.然后,使用得到的黄原酸化合物分散体,除此以外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
234.<比较例2>
235.在制备黄原酸化合物分散体时,没有进行破碎处理(即,仅单纯进行了混合),除此以外,与实施例1同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为14μm、95%体积累积径(d95)为55μm。
236.然后,使用得到的黄原酸化合物分散体,除此以外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
237.<比较例3>
238.(二乙基二硫代氨基甲酸锌的分散体的制备)
239.通过球磨机(商品名“瓷制球磨机”,日陶科学公司制)混合0.3份的二乙基二硫代氨基甲酸锌、0.9份的水和0.03份的作为非离子系表面活性剂的聚氧乙烯二苯乙烯化苯基醚(商品名“emulgen a
‑
60”,花王公司制)来进行破碎处理,由此得到二乙基二硫代氨基甲
酸锌的分散体。另外,利用球磨机的混合条件与实施例3相同。与实施例1同样地测定得到的二乙基二硫代氨基甲酸锌的分散体中的二乙基二硫代氨基甲酸锌的体积平均粒径和95%体积累积径(d50),结果二乙基二硫代氨基甲酸锌的体积平均粒径为1μm、95%体积累积径(d95)为4μm。
240.(二丁基二硫代氨基甲酸锌的分散体的制备)
241.通过球磨机(商品名“瓷制球磨机”,日陶科学公司制)混合0.5份的二丁基二硫代氨基甲酸锌、1.5份的水和0.05份的作为非离子系表面活性剂的聚氧乙烯二苯乙烯化苯基醚(商品名“emulgen a
‑
60”,花王公司制)来进行破碎处理,由此得到二丁基二硫代氨基甲酸锌的分散体。另外,利用球磨机的混合条件与实施例3相同。与实施例1同样地测定得到的二丁基二硫代氨基甲酸锌的分散体中的二丁基二硫代氨基甲酸锌的体积平均粒径和95%体积累积径(d50),结果二丁基二硫代氨基甲酸锌的体积平均粒径为3μm、95%体积累积径(d95)为13μm。
242.(2
‑
巯基苯并噻唑锌的分散体的制备)
243.将0.7份的2
‑
巯基苯并噻唑锌、2.1份的水和0.07份的作为非离子系表面活性剂的聚氧乙烯二苯乙烯化苯基醚(商品名“emulgen a
‑
60”,花王公司制)通过球磨机(商品名“瓷制球磨机”,日陶科学公司制)混合来进行破碎处理,由此得到2
‑
巯基苯并噻唑锌的分散体。另外,利用球磨机的混合条件与实施例3相同。与实施例1同样地测定得到的2
‑
巯基苯并噻唑锌的分散体中的2
‑
巯基苯并噻唑锌的体积平均粒径和95%体积累积径(d50),结果2
‑
巯基苯并噻唑锌的体积平均粒径为0.5μm、95%体积累积径(d95)为2μm。
244.(胶乳组合物的制备、浸渍成型体的制造)
245.然后,代替黄原酸化合物分散体而使用1.23份的上述制备的二乙基二硫代氨基甲酸锌的分散体(以二乙基二硫代氨基甲酸锌换算计为0.3部)、2.05份的二丁基二硫代氨基甲酸锌的分散体(以二丁基二硫代氨基甲酸锌换算计为0.5份)、和2.87份的2
‑
巯基苯并噻唑锌的分散体(以2
‑
巯基苯并噻唑锌计为0.7份),除此以外,与实施例1同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
246.<比较例4>
247.在制备二乙基二硫代氨基甲酸锌的分散体、二丁基二硫代氨基甲酸锌的分散体和2
‑
巯基苯并噻唑锌的分散体时,使利用球磨机的破碎处理的时间为24小时,除此以外,与比较例3同样地进行,得到二乙基二硫代氨基甲酸锌的分散体、二丁基二硫代氨基甲酸锌的分散体和2
‑
巯基苯并噻唑锌的分散体。得到的分散体中所包含的各硫化促进剂的体积平均粒径和95%体积累积径(d95)如表1所示。然后,使用得到的这些分散体,与比较例3同样地进行,得到48小时熟化胶乳组合物和72小时熟化胶乳组合物、以及浸渍成型体(48小时熟化品和72小时熟化品),同样地进行测定、评价。结果示于表1。
248.[表1]
[0249][0250]
<实施例1~5,比较例1~4的评价>
[0251]
如表1所示,使用含有体积平均粒径为0.001~9μm的黄原酸化合物的黄原酸化合物分散体得到的胶乳组合物提供迟发型过敏(type iv)的症状的产生被有效抑制的浸渍成型体,得到的浸渍成型体具有充分的撕裂强度,进而撕裂强度的稳定性也高,针孔的产生被有效抑制(实施例1~5)。
[0252]
另一方面,在使用含有体积平均粒径为10μm的黄原酸化合物的黄原酸化合物分散
体的情况下,结果得到的浸渍成型体会产生针孔,此外,在48小时的熟化时间下,撕裂强度低、撕裂强度的稳定性差,为了使这些特性充分,需要将熟化时间延长至非常长的72小时(比较例1)。
[0253]
此外,在使用含有体积平均粒径为14μm的黄原酸化合物的黄原酸化合物分散体的情况下,结果得到的浸渍成型体会产生针孔,此外,在使熟化时间为48小时、72小时的任一情况下,撕裂强度均低,撕裂强度的稳定性均差(比较例2)。
[0254]
进而,在代替黄原酸化合物使用二乙基二硫代氨基甲酸锌、二丁基二硫代氨基甲酸锌和2
‑
巯基苯并噻唑锌作为硫化促进剂的情况下,得到的浸渍成型体引起迟发型过敏(type iv)症状(比较例1、2)。另外,这些比较例1、2是使用体积平均粒径互不相同的硫化促进剂作为二乙基二硫代氨基甲酸锌、二丁基二硫代氨基甲酸锌和2
‑
巯基苯并噻唑锌的例子,结果比较例1、2两者的撕裂强度、撕裂强度的稳定性和针孔的抑制的效果为几乎相同程度。
[0255]
<实施例6>
[0256]
(黄原酸化合物分散体的制备)
[0257]
通过球磨机(商品名“瓷制球磨机”,日陶科学公司制)混合2份的作为黄原酸化合物的二异丙基黄原酸锌(商品名“nocceler zix”,大内新兴化学工业株式会社制,体积平均粒径:14μm,95%体积累积径(d95):55μm)、0.15份的作为非离子系表面活性剂的聚氧乙烯二苯乙烯化苯基醚(商品名“emulgen a
‑
60”,花王公司制)和4.5份的水来进行破碎处理,由此得到黄原酸化合物分散体。另外,作为利用球磨机的混合条件,使用和的陶瓷制球,以50rpm旋转72小时以上。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为3μm、95%体积累积径(d95)为16μm。
[0258]
(胶乳组合物的制备)
[0259]
首先,准备苯乙烯
‑
马来酸单仲丁酯
‑
马来酸单甲酯聚合物(商品名“scripset550”,hercules inc.制),使用氢氧化钠中和聚合物中的100%的羧基,由此制备钠盐水溶液(浓度10重量%)。然后,在制造例3中得到的羧基改性合成聚异戊二烯(a
‑
3)的胶乳中,以相对于胶乳中100份的羧基改性合成聚异戊二烯(a
‑
3)以固体成分换算计为0.8份的方式添加该钠盐水溶液,得到混合物。
[0260]
然后,一边搅拌得到的混合物,一边相对于混合物中100份的羧基改性合成聚异戊二烯(a
‑
3),添加6.65份的上述制备的黄原酸化合物分散体(以二异丙基黄原酸锌换算计为2份,以聚氧乙烯二苯乙烯化苯基醚换算计为0.15份)。
[0261]
接下来,以按固体成分换算计为1.5份的作为活化剂的氧化锌、1.5份的硫、2份的抗老化剂(商品名“wingstay l”、goodyear公司制)的方式,添加各添加剂的水分散液,得到胶乳组合物。然后,将得到的胶乳组合物分成2部分,对一部分用调节至25℃的恒温水槽进行6小时熟化(前硫化),对另一部分用调节至25℃的恒温水槽进行48小时熟化(前硫化),由此得到6小时熟化胶乳组合物和48小时熟化胶乳组合物。
[0262]
然后,使用得到的6小时熟化胶乳组合物和48小时熟化胶乳组合物,除此以外,与实施例1同样地进行,得到手套形状的浸渍成型体(6小时熟化品和48小时熟化品),与实施例1同样地进行,同样地进行测定、评价。结果示于表2。
[0263]
<实施例7>
[0264]
代替制造例3中得到的羧基改性合成聚异戊二烯(a
‑
3)的胶乳,使用制造例2中得到的羧基改性合成聚异戊二烯(a
‑
2)的胶乳,除此以外,与实施例6同样地进行,得到了6小时熟化胶乳组合物和48小时熟化胶乳组合物、以及浸渍成型体(6小时熟化品和48小时熟化品),同样地进行测定、评价。结果示于表2。
[0265]
<实施例8>
[0266]
在制备黄原酸化合物分散体时,作为黄原酸化合物,代替2份的二异丙基黄原酸锌而使用2份的丁基黄原酸锌,除此以外,与实施例6同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的丁基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果丁基黄原酸锌的体积平均粒径为3μm、95%体积累积径(d95)为16μm。
[0267]
然后,使用得到的黄原酸化合物分散体,除此以外,与实施例6同样地进行,得到6小时熟化胶乳组合物和48小时熟化胶乳组合物、以及浸渍成型体(6小时熟化品和48小时熟化品),同样地进行测定、评价。结果示于表2。
[0268]
<实施例9>
[0269]
在制备黄原酸化合物分散体时,作为黄原酸化合物,代替2份的二异丙基黄原酸锌而使用2份的乙基黄原酸锌,除此以外,与实施例6同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的乙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果乙基黄原酸锌的体积平均粒径为3μm、95%体积累积径(d95)为16μm。
[0270]
然后,使用得到的黄原酸化合物分散体,除此以外,与实施例6同样地进行,得到6小时熟化胶乳组合物和48小时熟化胶乳组合物、以及浸渍成型体(6小时熟化品和48小时熟化品),同样地进行测定、评价。结果示于表2。
[0271]
<实施例10>
[0272]
在制备黄原酸化合物分散体时,作为非离子系表面活性剂,代替0.15份的聚氧乙烯二苯乙烯化苯基醚而使用0.15份的聚氧乙烯月桂基醚(聚氧乙烯(6)月桂基醚,商品名“emulgen 108”,花王公司制),除此以外,与实施例6同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为3μm、95%体积累积径(d95)为16μm。
[0273]
然后,使用得到的黄原酸化合物分散体,除此以外,与实施例6同样地进行,得到6小时熟化胶乳组合物和48小时熟化胶乳组合物、以及浸渍成型体(6小时熟化品和48小时熟化品),同样地进行测定、评价。结果示于表2。
[0274]
<实施例11>
[0275]
在制备黄原酸化合物分散体时,作为非离子系表面活性剂,代替0.15份的聚氧乙烯二苯乙烯化苯基醚而使用0.15份的聚氧乙烯烷基醚(聚氧乙烯(9)烷基(仲c11
‑
15)醚,商品名“emulgen 709”,花王公司制),除此以外,与实施例6同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为3μm、95%体积
累积径(d95)为16μm。
[0276]
然后,使用得到的黄原酸化合物分散体,除此以外,与实施例6同样地进行,得到6小时熟化胶乳组合物和48小时熟化胶乳组合物、以及浸渍成型体(6小时熟化品和48小时熟化品),同样地进行测定、评价。结果示于表2。
[0277]
<实施例12>
[0278]
在制备黄原酸化合物分散体时,作为非离子系表面活性剂,代替0.15份的聚氧乙烯二苯乙烯化苯基醚而使用0.15份的聚氧乙烯聚氧丙二醇(商品名“emulgen pp
‑
290”,花王公司制),除此以外,与实施例6同样地进行,得到黄原酸化合物分散体。与实施例1同样地测定得到的黄原酸化合物分散体中的二异丙基黄原酸锌的体积平均粒径和95%体积累积径(d95),结果二异丙基黄原酸锌的体积平均粒径为3μm,95%体积累积径(d95)为16μm。
[0279]
然后,使用得到的黄原酸化合物分散体,除此以外,与实施例6同样地进行,得到6小时熟化胶乳组合物和48小时熟化胶乳组合物、以及浸渍成型体(6小时熟化品和48小时熟化品),同样地进行测定、评价。结果示于表2。
[0280]
[表2]
[0281][0282]
<实施例6~12的评价>
[0283]
如表2所示,使用含有体积平均粒径为0.001~9μm的黄原酸化合物的黄原酸化合物分散体得到的胶乳组合物提供迟发型过敏(type iv)症状的产生被有效抑制的浸渍成型体,得到的浸渍成型体具有充分的撕裂强度,进而撕裂强度的稳定性也高,针孔的产生被有效抑制(实施例6~12)。此外,实施例6~12的黄原酸化合物分散体均含有非离子系表面活性剂和/或非离子阴离子系表面活性剂,根据实施例6~12的结果可知,即使在将熟化缩短
至6小时的情况下,也能够得到具有充分的机械强度的浸渍成型体,根据该结果,确认了通过进一步含有非离子系表面活性剂和/或非离子阴离子系表面活性剂,能够缩短熟化(前硫化)所需要的时间,生产率优异。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1