用于管理下一代测序中的低样本输入的归一化对照的制作方法
相关申请
本申请要求于2018年10月4日提交的美国临时申请号62/741,466和2019年4月11日提交的美国临时申请号62/832,560的优先权和权益,所述美国临时申请中的每一者的内容以全文引用方式并入本文。
本发明涉及使用高通量测序对样本中的核酸进行定量的方法和组合物。
通过引用并入序列表
本申请与电子格式的序列表一起提交。序列表作为名为arcb-006-02wo_seqlist.txt的文件提供,创建于2019年10月3日,并且大小为5943字节。序列表的电子格式信息以引用方式整体并入。
背景技术:
下一代测序(nextgenerationsequencing,ngs),也称为高通量测序,是用于研究和诊断的强大工具。ngs技术允许大规模的核酸高通量测序。使用ngs技术,可以廉价高效地对来自单个样本的数百万个dna或rna分子(多核苷酸)进行测序。在ngs测序期间取样多核苷酸的程度与样本中多核苷酸的初始浓度成正比。因此,ngs可用于确定测序样本中单个多核苷酸或多核苷酸群体的数量。然而,由ngs生成的读段数量和样本中多核苷酸的起始浓度不一定很好地相关。当从低样本输入开始时,使ngs读段数和起始浓度相关就特别成问题,因为样本制备和测序期间的随机过程在起始样本输入低时会对ngs过程中测序的核酸分子的数量具有不成比例的影响。在各种临床应用中,例如,当尝试检测样本中以低滴度存在的病原体时,起始样本输入量可能很低。
在本领域中需要在使用ngs时准确定量样本中多核苷酸的丰度的组合物和方法。本文提供了可用于使用ngs对样本中的多核苷酸进行这种定量的方法和组合物。
技术实现要素:
本文提供了用于下一代测序的归一化对照、所述归一化对照的制备方法,以及使用所述归一化对照的方法。
在一个方面中,本文提供了归一化对照,所述归一化对照包括至少三组多核苷酸,其中每个组内的多核苷酸具有相同的长度。在一些实施方式中,所述归一化对照中的所有多核苷酸的长度是相同的。在一些实施方式中,每个组内的多核苷酸当与任何其他组内的多核苷酸相比时具有不同的长度。
在本公开的归一化对照的一些实施方式中,每个组内的多核苷酸包含相同的序列。在一些实施方式中,每个组内的多核苷酸不包含相同的序列。在一些实施方式中,每组多核苷酸包含多核苷酸的至少三个亚组,其中每个亚组内的多核苷酸包含相同的序列,并且其中每个亚组内的多核苷酸不包含与任何其他亚组相同的序列。
在本公开的归一化对照的一些实施方式中,至少一组多核苷酸的序列包含组分序列。在一些实施方式中,每组多核苷酸的序列包含组分序列。在一些实施方式中,组分序列包含随机序列或由随机序列组成。
在本公开的归一化对照的一些实施方式中,至少一组多核苷酸的序列包含分离的序列。在一些实施方式中,每组多核苷酸的序列包含分离的序列。在一些实施方式中,分离的序列是分离的或或来源于病毒、细菌、真菌或真核寄生虫。在一些实施方式中,分离的序列与测序样本中的靶序列不同。
在本公开的归一化对照的一些实施方式中,归一化对照中的至少一个亚组或至少一组多核苷酸的序列与测序样本中的靶序列共享至少一种序列特性。所述至少一个序列特性是例如转座元件序列、病毒序列、细菌序列、真菌序列、真核寄生虫序列、或一个或多个人类基因序列的序列特性。
在本公开的归一化对照的一些实施方式中,所述测序样本包含宿主核酸和非宿主核酸的混合物。在一些实施方式中,宿主是真核生物,例如昆虫、植物或动物。在一些实施方式中,宿主是人。在一些实施方式中,非宿主包含共生体、共生生物、寄生虫或病原体。非宿主可以包含多个物种。在一些实施方式中,靶序列是测序样本中的非宿主序列。在示例性实施方式中,非宿主是病毒、细菌或真菌,并且靶序列包含非宿主的毒力因子的序列。
在另一方面中,本文提供了制备归一化对照的方法,所述方法包括:(a)合成至少三组dna分子,其中一组内的dna分子具有相同的序列,并且其中所述dna分子中的每个dna分子的序列从5'至3'包含第一组分序列、第一限制性酶切位点、第二组分序列、第二限制性位点和第三组分序列;(b)对溶液中各组dna分子中的每个组的量进行定量;(c)用切割第一和第二限制位点的限制性酶消化所述多组dna分子中的每个组,以产生至少三组经消化的dna分子;以及(d)将预定量的所述多组经消化的dna分子中的各个组混合,从而产生归一化对照。
在另一方面中,本文提供了制备包含rna分子的归一化对照的方法,所述方法包括:(a)合成至少三组dna分子,其中所述至少三组dna分子中的每一组从5'至3'包含第一组分序列、启动子序列、第一限制性酶切位点、第二组分序列、第二启动子序列、第二限制性位点、第三组分序列和第三启动子序列;(b)用切割第一和第二限制性酶切位点的限制性酶消化每组dna分子,以产生至少三组消化的dna分子;(c)在体外转录所述至少三组经消化的dna分子中的每个组,以产生至少三个rna分子群体;(d)对所述rna分子群体中的每个rna分子群体中产生的rna的量进行定量;以及(e)混合预定量的所述rna分子群体中的每个rna分子群体以产生包含rna分子的归一化对照。
在另一方面中,本文提供了制备包含至少三组多核苷酸的归一化对照的方法,其中每个组内的多核苷酸具有相同的长度,所述方法包括:(a)从样本中提取dna;(b)用限制性酶消化dna以产生dna片段集合;(c)分离所述dna片段集合;(d)纯化dna片段以产生至少三组多核苷酸,其中每个组内的多核苷酸具有相同的长度;以及(e)混合预定量的所述各组多核苷酸中的每个组以产生归一化对照。
在另一方面中,本文提供了对样本中至少一个靶核酸分子的表达水平进行定量的方法,所述方法包括:(a)将已知量的本公开的归一化对照与样本混合,(b)制备高通量测序文库,(c)对所述文库测序以产生读段集合,(d)将每个读段映射至样本或归一化对照,(e)确定由归一化对照中的所述组或亚组多核苷酸中的每个组或亚组产生的读段的数量,(f)计算(a)中与样本混合的归一化对照中的多种核酸分子中的每种核酸分子的起始浓度与(c)中产生的读段的数量之间的关系,(g)确定读段与样本中浓度之间的关系,以及(h)使用(g)中的模型根据至少一个靶核酸分子产生的读段的数量计算样本中所述靶核酸分子的初始浓度。
本文还提供了用于下一代测序的多分析物对照、所述多分析物对照的制备方法,以及使用所述多分析物对照的方法。在一些实施方式中,本文所述的归一化对照用于归一化来自多分析物对照和样本的读段,并且归一化读段用于确定样本中的一种或多种靶生物体的滴度。
在一个方面中,本文提供了包含至少三种不同物种的生物体的混合物的多分析物对照,其中所述生物体已被灭活。在一些实施方式中,多分析物对照包含至少十种不同物种的生物。
在另一方面中,本文提供了对样本中至少一个靶序列进行定量的方法,其中所述样本包含多分析物对照,所述对照包含至少三种不同物种的生物体的混合物,其中所述生物体已被灭活。
在另一方面中,本文提供了对样本中的至少一个靶序列进行定量的方法,所述方法包括将已知量的多分析物对照与样本混合,其中所述多分析物对照包括至少三种不同物种的生物体的混合物,并且其中所述生物体已被灭活。
在另一方面,本文提供了对样本中的靶生物体体的滴度进行定量的方法,所述方法包括:(a)提供包含所述靶生物体体的样本,其中所述靶生物体体包含至少一个靶序列;以及(b)提供包含已知滴度的所述至少三种物种的生物体的多分析物对照,其中所述生物体已被灭活;(c)将已知量的本公开的归一化对照物与样本和多分析物对照混合;(d)从样本和多分析物对照制备高通量测序文库;(e)对所述文库测序以产生样本读段集合和多分析物对照读段集合;(f)使用归一化对照对来自(e)的样本读段集合和多分析物对照读段集合进行归一化;(g)确定归一化读段与来自多分析物对照中的至少三种物种的生物体的生物体的已知滴度之间的关系;以及(h)使用(g)中确定的关系计算样本中的靶生物体体的滴度。
在一些实施方式中,步骤(f)处的归一化包括:(i)将来自样本读段集合的读段映射至样本或归一化对照;(ii)将来自多分析物对照读段集合的读段映射至多分析物对照或归一化对照;(iii)针对样本读段集合和多分析物对照读段集合确定归一化对照中的所述组或亚组多核苷酸中的每个组或亚组产生的读段的数量;(iv)计算归一化对照中的所述组或亚组多核苷酸中的每个组或亚组的起始浓度与在步骤(e)中产生的样本和多分析物对照两者的读段的数量之间的关系;以及(v)确定样本和多分析物对照中的读段与浓度之间的关系。
本公开提供了制备多个归一化对照寡核苷酸的方法,所述方法包括:(a)从至少一个参考序列产生多个参考序列片段;(b)根据参考序列片段的数量产生至少一个参数的分布;(c)将所述分布划分成至少5个箱;(d)从至少5个箱中的至少3个箱中选择至少一个参考序列片段;(e)改组至少3个参考序列以产生经改组的序列;以及(f)合成包含经改组的序列的寡核苷酸;从而产生多个归一化对照寡核苷酸。
本公开提供了制备多个归一化对照寡核苷酸的方法,所述方法包括:(a)使用滑动窗从至少一个参考序列产生多个参考序列片段;(b)根据参考序列片段的数量产生至少一个参数的分布;(c)将所述分布划分为至少5个箱;(d)从至少5个箱中的至少3个箱中选择至少两个参考序列片段,其中所述至少两个参考序列片段在参考序列中是不连续的,或者是从不同的参考序列中选择的;(e)将来自至少3个箱中的每一个箱的至少两个参考序列片段连接起来;以及(f)合成包含连接的参考序列片段的寡核苷酸;从而产生多个归一化对照寡核苷酸。在一些实施方式中,根据权利要求203-206中任一项所述的方法,其中所述滑动窗口包括1bp、2bp、3bp、4bp或5bp的滑动窗口。在一些实施方式中,所述参考序列片段是对应参考序列的约15-60个、约20-40个、约20-30个、约15-32个、20-32个或约25-35个连续核苷酸。在一些实施方式中,参考序列片段是对应参考序列的29个、30个、31个、32个、33个或34个连续核苷酸。
在本公开的方法的一些实施方式中,所述参数包括以下各项中的至少一项:(1)gc含量百分比、(2)熵、(3)复杂性、(4)eiip,或它们的组合。
在本公开的方法的一些实施方式中,所述至少一个参考序列包含至少2个、至少10个、至少20个、至少50个、至少100个、至少200个、至少300个、至少400个、至少500个、至少600个、至少700个、至少800个、至少900个、至少1,000个、至少1,200个、至少1,300个、至少1,400个、至少1,500个、至少1,600个、至少1,700个、至少1,800个、至少1,900个、至少2,000个、至少2,200个、至少2,400个、至少2,600个、至少2,800个、至少3,000个、至少4,000个、至少5,000个、至少6,000个、至少7,000个、至少8,000个、至少9,000或至少10,000个参考序列。在一些实施方式中,至少一个参考序列包含约2,000个参考序列。
在本公开内容的方法的一些实施方式中,每个归一化对照寡核苷酸具有以下中的至少三者:与来自参考序列分布中的对应箱的参考序列片段(1)类似的gc含量百分比、(2)类似的熵、(3)类似的eiip、(4)类似的长度、或(5)类似的复杂性。
本公开提供了富集样本中感兴趣的靶核酸以供与本文提供的归一化对照和多分析物对照组合使用的方法。在一些实施方式中,所述方法包括基于核苷酸修饰的富集方法。在一些实施方式中,所述方法包括基于核酸引导核酸酶的富集方法。在一些实施方式中,所述方法包括基于核苷酸修饰的富集方法和基于核酸引导核酸酶的富集方法两者。
在本公开的方法的一些实施方式中,富集样本中感兴趣的核酸的方法包括:(a)提供包含感兴趣的核酸和靶向耗尽的核酸的样本,其中所述感兴趣的核酸的至少一个子集或所述靶向耗尽的核酸的子集包含用于第一修饰敏感的限制性酶的多个第一识别位点;(b)使样本中的多种核酸末端去磷酸化;(c)在允许切割样本中的核酸中的第一修饰敏感限制性位点中的至少一些第一修饰敏感限制性位点的条件下,使来自(b)的样本与第一修饰敏感限制性酶接触;以及(d)在允许衔接子与多个感兴趣的核酸的5'末端和3'末端连接的条件下,使来自(c)的样本与衔接子接触;从而产生富集感兴趣的核酸的样本,所述感兴趣的核酸在其5'末端和3'末端与衔接子连接。
在本公开的方法的一些实施方式中,所述方法使样本与多种核酸引导的核酸酶引导核酸(nuclease-guidenucleicacid,gna)复合物接触,其中所述gna与靶向耗尽的核酸中的靶位点互补,从而产生对在一个末端处与衔接子连接的靶向耗尽的核酸和在5'末端和3'末端处均与衔接子连接的感兴趣的靶核酸的切割。在一些实施方式中,该方法包括使样本与至少102个独特的核酸引导的核酸酶-gna复合物、至少103个独特的核酸引导的核酸酶-gna复合物、104个独特的核酸引导的核酸酶-gna复合物、或105个独特的核酸引导的核酸酶-gna复合物接触。在一些实施方式中,核酸引导的核酸酶是cas9、cpf1或它们的组合。
在其他方面中,本文提供了包含本公开的多分析物对照的试剂盒、包含本公开的归一化对照的试剂盒,以及包含本公开的归一化对照和多分析物对照的试剂盒。在一些实施方式中,试剂盒还包含用于感兴趣的靶核酸的基于核苷酸修饰的富集的试剂、用于感兴趣的靶核酸的基于核酸引导核酸酶的富集的试剂,或两者。
本公开提供了包含本公开的归一化对照的试剂盒。
本公开提供了包含本公开的多分析物对照的试剂盒。
本公开提供了试剂盒,所述试剂盒包含归一化对照、多分析物对照、试剂和使用说明书。在一些实施方式中,试剂盒还包含用于耗尽靶向耗尽的序列的试剂和使用说明书。
本公开提供了用于设计多个归一化对照多核苷酸序列的系统。在一些实施方式中,该系统包括计算机可读存储介质,该计算机可读存储介质存储计算机可执行指令,该计算机可执行指令包括:(i)用于导入至少一个参考序列的指令;(ii)从至少一个参考序列产生多个参考序列片段的指令;(iii)根据参考序列片段数目产生至少一个参数的分布的指令;(iv)将分布划分成箱的指令;(v)从箱的至少一个子集中选择多个参考序列片段的指令;和(vi)改组多个参考序列片段以产生改组序列的指令;从而产生多个归一化多核苷酸序列。在一些实施方式中,该系统包括计算机可读存储介质,该计算机可读存储介质存储计算机可执行指令,该计算机可执行指令包括:(i)用于导入至少一个参考序列的指令;(ii)从至少一个参考序列产生多个参考序列片段的指令;(iii)根据参考序列片段数目产生至少一个参数的分布的指令;(iv)将分布划分成箱的指令;(v)从箱的至少一个子集中的每一个中选择至少两个参考序列片段的指令,其中所述至少两个参考序列片段在参考序列中是不连续的,或来自不同的参考序列;(vi)连接来自每个箱的至少两个参考序列片段的指令;从而产生多个归一化多核苷酸序列。在一些实施方式中,该系统还包括处理器,该处理器被配置成执行包括以下步骤的步骤:(a)接收包括至少一个参考序列的一组输入文件;和(b)执行存储在计算机可读存储介质中的计算机可执行指令。在一些实施方式中,该参数包括以下各项中的至少一项:(1)gc含量百分比、(2)熵、(3)复杂性、(4)eiip、(5)长度,或它们的组合。
附图说明
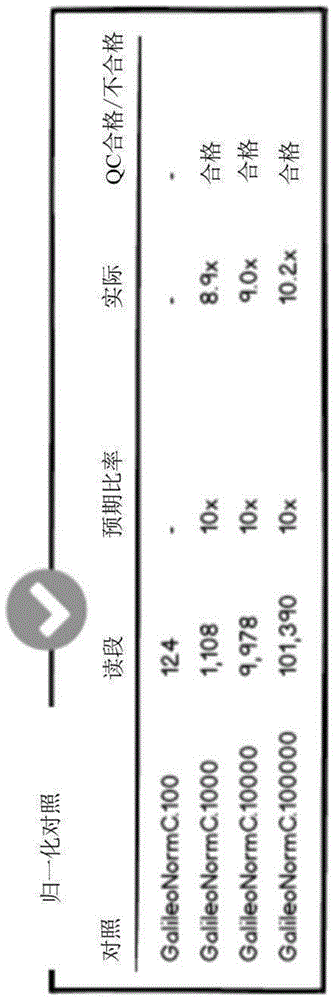
图1是示出归一化对照(normalizationcontrol,nc)读段的质量控制分析的表。
图2是绘制巨细胞病毒(cytomegalovirus,cmv)丰度(x轴)对比滴度(y轴)的图。如果不进行归一化,则r2值为0.89。
图3是绘制归一化对照(nc)归一化的cmv丰度(x轴)对比滴度(y轴)的图。通过归一化,r2值为0.98。
图4是条形图,示出了用三个假想的下一代测序文库分析的样本中病毒的丰度百分比(y轴,任意单位),以证明使用不同转化计算得出的病毒的丰度百分比。对于每个文库,条形从左到右分别代表已知的病毒载量、使用下一代测序后的原始读段迹象计算的病毒载量以及使用f(r)读段证据和f(r)+δr读段迹象计算的病毒载量。
图5是示出当使用f(r)计算病毒载量对比使用原始读段计算病毒载量时预期的可变性降低的图。
图6是用于定量样本中靶序列水平的方案的概述,其中在rna提取过程中添加归一化对照。
图7是示出精确诊断阴性血浆样本文库中归一化对照读段百分比的图,其中在样本提取期间以低、中和高量向样本中添加nc。
图8是用于将双索引引物与归一化对照退火的方案的表。
图9是示出在添加归一化对照的情况下文库中的归一化对照读段百分比的图,其中dnanc以高(左两栏)、中(右两栏)和低(中两栏)的量添加。
图10是示出示例性归一化对照的gc含量、总计数和长度的表。
图11是示出本公开的示例性归一化对照的浓度和比率的表。从左至右,各列示出归一化对照组合物中的各组多核苷酸(在图11中称为“寡核苷酸”,并具有对应的寡核苷酸编号)、每组多核苷酸的最终浓度(ng/μl),以及对于组合物中归一化对照多核苷酸的群体,归一化对照组合物中特定组的多核苷酸的dna的量与归一化对照组合物中所有多核苷酸的所有dna总量之比。
图12是示出本公开的示例性rna归一化对照的大小、输入分子数量和浓度的表。从左至右,各列示出了归一化对照中的单独rna片段、归一化对照中的各组rna、rna分子的长度(以kb和bp为单位)、输入分子的数量、每个rna分子在其原始gblock中的阿摩尔(attomole)数,以及在gblock片段化后每个rna分子的所得浓度(以阿摩尔/微升计)。
图13是示出以log为单位的滴度作为用于校准和实验样本的观察信号的函数的图。深灰色圆圈表示用于校准的多分析物对照样本中的物质,而浅灰色圆圈表示实验样本。
图14a是示出用于设计本公开的归一化对照的示例性过程的流程图。
图14b是示出本公开的归一化对照的一种设计的信号(y轴)和对数浓度(x轴)的图。
图14c是示出本公开的一组归一化对照的浓度的表。每个寡核苷酸是不同的序列。
图14d是示出本公开的归一化对照的一种设计的信号(y轴)和对数浓度(x轴)的图。浓度的每个数量级由6个浓度表示,每个对数浓度具有至少一个不同的寡核苷酸,并且覆盖了7-8个数量级。
图15是示出关于大约1200个参考基因组的熵、电子-离子相互作用电势(electron-ioninteractionpotential,eiip)和gc含量之间的相互作用的三元图。
图16是一系列三个曲线图,示出了来自使用改组方法产生的参考序列的kmer的分布。从左至右:电子eiip、熵和gc含量。eiip、熵和gc含量的值分别在x轴上从左至右示出。在y轴上示出了kmer密度。该线表示分布,并且直方图条表示给定箱中kmer数量的实际计数。
图17a是一系列三个曲线图,示出了使用改组方法产生的1000万个kmer的分布。从左至右:电子eiip、熵和gc含量。eiip、熵和gc含量的值分别在x轴上从左至右示出。在y轴上示出了kmer密度。
图17b是一系列三个曲线图,示出了使用改组方法产生的130万个kmer的分布。从左至右:电子eiip、熵和gc含量。eiip、熵和gc含量的值分别在x轴上从左至右示出。在y轴上示出了kmer密度。
图18是示出在改组之前将kmer连接以产生归一化对照序列的过程的图。
图19a是一系列三个图,示出了使用cantor改组方法生成的归一化对照与bk多瘤病毒分离株ch-1之间的eiip(左上)、gc含量(右上)和复杂性(底部行)对应关系。未改组的kmer绘制在x轴上,机构cantor改组的kmer绘制在y轴上,线表示1:1对应。
图19b是一系列三个图,示出了使用cantor改组方法生成的归一化对照与jc多瘤病毒株niid12-31之间的eiip(左上)、gc含量(右上)和复杂性(底部行)对应关系。未改组的kmer绘制在x轴上,机构cantor改组的kmer绘制在y轴上,线表示1:1对应。
图19c是一系列三个图,示出了使用cantor改组方法产生的归一化对照与人多瘤病毒1菌株bk2之间的eiip(左上)、gc含量(右上)和复杂性(底部行)对应关系。未改组的kmer绘制在x轴上,机构cantor改组的kmer绘制在y轴上,线表示1:1对应。
图19d是一系列三个图,示出了使用cantor改组方法生成的归一化对照与的人腺病毒c之间的eiip(左上)、gc含量(右上)和复杂性(底部行)对应关系。未改组的kmer绘制在x轴上,机构cantor改组的kmer绘制在y轴上,线表示1:1对应。
图20a是基本局部比对搜索工具(basiclocalalignmentsearchtool,blast)比对,示出了使用ncbiblast核苷酸序列时,经cantor改组的归一化对照序列不与bk多瘤病毒分离株ch-1进行比对。
图20b是blast比对,示出了使用ncbiblast核苷酸序列时,经cantor改组的归一化对照序列不与jc多瘤病毒株niid12-31比对。
图20c是blast比对,示出了使用ncbiblast核苷酸序列时,经cantor改组的归一化对照序列不与人多瘤病毒1菌株bk2比对。
图20d是blast比对,示出了使用ncbiblast核苷酸序列时,经cantor改组的归一化对照序列不与人腺病毒c比对。
图21a是一系列三个曲线图,示出了具有选定的gc含量百分比的kmer的eiip、熵和gc含量。
图21b是一系列曲线图,示出了具有选定的gc含量百分比的kmer的eiip、熵和gc含量。
图21c是一系列曲线图,示出了具有选定的gc含量百分比的kmer的eiip、熵和gc含量。
图22是一对曲线图,示出了eipp、gc含量和熵的第一和第二主分量(分别为x轴和y轴)。计算了pc1和pc2的参考基因组序列(左)和使用滑动窗口/穷举kmer方法生成的归一化对照序列(右)。
图23是一系列三个q-q曲线图,比较了参考基因组序列(x轴)和使用滑动窗/穷举kmer方法生成的归一化对照寡核苷酸(y轴)的gc含量(顶部)、eiip(左下)和熵(右下)。
图24是kolmogorov-smirnov(ks)检验曲线图,比较了参考基因组序列和使用滑动窗/穷举kmer方法生成的归一化对照序列的随机子取样群体中的熵。ecdf=经验累积分布函数。箭头指示归一化对照的熵。点表示k-s统计。
图25a是blast比对,示出了使用滑动窗/穷举kmer方法生成的785,000个归一化对照序列的文件头部的序列在约750个碱基对(bp)的位置与假单胞菌属(pseudomonas)的约50-80bp比对。
图25b是blast比对,示出了使用blast时,使用滑动窗/穷举kmer方法生成的785,000个归一化对照序列的文件尾部的序列不与任何序列进行比对。
图26是比较使用改组和穷举kmer/滑动窗方法设计的归一化对照的表。
图27是示出本发明的示例性方法的图。样本中的核酸被去磷酸化,然后用限制性酶消化,该限制性酶通过限制性酶识别位点处的修饰的存在而被封端。然后将来自所得消化的暴露的磷酸酯用于将衔接子连接至感兴趣的核酸。
图28是示出本发明的示例性方法的图。将样本中的核酸去磷酸化,然后用识别限制性酶切位点的限制性酶消化,该限制性酶切位点包含一个或多个经修饰的核苷酸。然后,将切出的核酸用核酸外切酶消化,所述核酸外切酶使用暴露的末端磷酸酯,并将衔接子与其余的感兴趣的核酸连接。
图29是示出本发明的示例性方法的图。将样本中的核酸与衔接子连接,然后用限制性酶消化,所述限制性酶识别包含一个或多个经修饰的核苷酸的限制性酶切位点,从而产生在两个末端处均与衔接子连接的感兴趣的核酸。
图30是示出了本公开的示例性方法的图。将样本中的核酸与衔接子连接,然后用核酸引导的核酸酶进行切割,该核酸引导的核酸酶切割靶向耗尽的核酸,从而产生在两个末端上均与衔接子连接的感兴趣的核酸。该方法可以与本公开的基于核苷酸修饰的方法结合使用。
具体实施方式
本文提供了用作下一代测序(nextgenerationsequencing,ngs)中样本的定量的归一化对照的组合物。在一些实施方式中,当使用低样本输入工作以进行ngs时使用归一化对照。在一些实施方式中,归一化对照用于计算样本中生物体的滴度,例如使用如本文所述的多分析物对照。在一些实施方式中,如本文所述,将归一化对照与富集样本中的靶序列的方法组合。
本文提供了制备归一化对照的方法,以及在ngs期间使用归一化对照的方法。
除非本文另外定义,否则本文使用的所有技术术语和科学术语的含义与如本公开所属领域的普通技术人员通常所理解的含义相同。尽管在本公开的实践或测试中可以使用类似于或等同于本文所述的方法和材料的任何方法和材料,但是描述了示例性方法和材料。
术语“下一代测序”(ngs)是指所谓的并行合成测序或连接测序平台,例如,目前由illumina、lifetechnologies和roche采用的那些平台。下一代测序方法还可以包括纳米孔测序方法或基于电子检测的方法,例如由lifetechnologies商售的iontorrent技术。ngs在本文中也称为“高通量测序”。
“测序样本”是包含使用ngs测序的核酸的样本。核酸可以是脱氧核糖核酸(deoxyribonucleicacid,dna)、核糖核酸(ribonucleicacid,rna)或两者。
“靶序列”是指测序样本中的单独序列或序列组,所述样本中的丰度是通过使用本公开的nc在ngs反应中测定的。靶序列的丰度可以为非零(即,靶序列以一定的丰度存在于样本中)或零(即,不存在靶序列或低于检测水平)。
如本文所用,“参考序列”是指与任何已知靶序列相同或相似的一个或多个序列。靶序列可以包含一个参考序列或多个参考序列或由其组成。
“靶生物体”是指样本中包含靶序列的生物体。
如本文所用,术语“组分序列”是指在归一化对照中发现的多核苷酸的一部分或全部。归一化对照多核苷酸包含组分序列或由其组成。可以从天然存在的序列、经工程化的质粒或合成物中分离组分序列。术语“分离的序列”是指已经分离或来源于生物体或其他先前存在的序列的类型的组分序列。分离的序列可以类似于它们所分离自或来源于的生物体的序列,或者也可以经受一种或多种转化,例如改组或连接。
术语“低样本”或“低输入样本”是指包含比常规文库制备方案中使用的核酸量低的核酸量的样本。低样本输入量可包含样本中总核酸中的至多100ng的核酸,以及少至1皮克核酸。在某些实施方式中,术语“低样本”或“低样本输入”是指编码样本中的一个或多个靶序列的核酸的量。低样本输入可包含至多100ng包含靶序列的核酸,或少于1飞克的包含靶序列的核酸。对于特别罕见的靶序列,飞摩尔和阿摩尔(attomolar)范围内的靶序列的浓度被认为在本公开的范围内。
如在整个本公开中所使用的,“序列同一性”或“序列相似性”可以通过使用默认参数,使用用于对两个序列进行blast的独立可执行blast引擎程序(bl2seq)来确定,所述程序可以从国家生物技术信息中心(nationalcenterforbiotechnologyinformation,ncbi)ftp站点取得(tatusova和madden,femsmicrobiollett.,1999,174,247-250;该文献以引用方式整体并入本文)。当在两个或更多个核酸或多肽序列的情境中使用时,术语“同一”或“同一性”是指在每个序列的指定区域上相同的残基的指定百分比。可以通过以下方法计算出百分比:对两个序列进行最佳比对;在指定区域内比较两个序列;确定两个序列中出现相同残基的位置数,以得到匹配的位置数;将匹配的位置数除以指定区域中的位置总数;以及将结果乘以100以得出序列同一性百分比。在两个序列具有不同的长度。或者比对产生一个或多个交错的末端并且指定的比较区域仅包含单个序列的情况下,单个序列的残基包含在计算的分母,而不是分子中。比较dna和rna时,胸腺嘧啶(t)和尿嘧啶(u)可以认为是等效的。同一性可以手动执行,或通过使用计算机序列算法(例如blast或blast2.0)执行。
如果通过比对序列是至少50%、至少60%、至少70%、至少75%、至少80%、至少85%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%、至少99%、至少99.5%或至少99.9%同一的,则所述序列是“类似的”。
本公开提供了要添加到用于ngs的样本中的归一化对照。任选地,该样本是低输入样本。
本公开提供了制备归一化对照的方法以及使用归一化对照的方法。
在一些实施方式中,将归一化对照以相同的量添加至每个样本中。
在一些实施方式中,不将归一化对照以相同的量添加至每个样本中。
在一些实施方式中,在核酸提取之前将归一化对照添加至样本中。在其他实施方式中,在核酸提取之后和文库制备期间添加归一化对照。在其他替代方案中,在文库制备之后和测序之前将归一化对照添加到样本中。
本公开提供了包含至少三组多核苷酸的归一化对照,其中所述至少三组中的每个组内的多核苷酸具有相同的长度。在一些实施方式中,归一化对照多核苷酸包含组分序列,在一些实施方式中,所述组分序列被设计为类似于或概括待测序的靶样本的一些特征或特性。在一些实施方式中,组分序列是随机序列。在一些实施方式中,归一化对照多核苷酸包含分离的序列。在一些实施方式中,归一化对照多核苷酸包含分离的序列和组分序列两者。在一些实施方式中,归一化对照多核苷酸的序列与测序样本中的靶序列共享至少一种序列特性。
本公开提供了制备归一化对照的方法,所述方法包括:(a)合成至少三个dna分子群体,其中一个群体内的每个dna分子具有相同的序列,并且其中所述dna分子中的每个dna分子的序列从5'至3'包含第一组分序列、第一限制性酶切位点、第二组分序列、第二限制性位点和第三组分序列;(b)对溶液中dna分子群体中的每个群体的量进行定量;(c)用切割第一和第二限制位点的限制性酶消化所述dna分子群体中的每个群体,以产生至少三个经消化的dna分子群体;以及(d)将预定量的所述经消化的dna分子群体中的各个群体在单个归一化对照组合物中混合。
在一些实施方式,例如其中样本是rna样本并且归一化对照是rna归一化对照的那些实施方式中,本公开提供了制备归一化对照的方法,所述方法包括:(a)合成具有至少三个dna分子的群体;其中所述至少三个合成dna分子中的每一个dna分子从5'至3'包含第一组分序列、与启动子序列互补的序列、第一限制性酶切位点、第二组分序列、与启动子序列互补的第二序列、第二限制性位点、第三组分序列和与启动子序列互补的第三序列;(b)用切割第一和第二限制性酶切位点的限制性酶消化每个dna分子群体,以产生至少三个经消化的dna分子群体;(c)在体外转录所述至少三个经消化的dna分子群体中的每一个群体,以产生至少三个rna分子群体;(d)对每个rna分子群体中产生的rna数量进行定量;以及(e)混合预定量的各个rna分子群体以产生单一的归一化对照组合物。
本公开提供了制备归一化对照的方法,所述方法包括:(a)从样本中提取dna;(b)用限制性酶消化dna以产生dna片段集合;以及(c)分离所述dna片段集合;(d)纯化至少三个dna片段以产生至少三组多核苷酸,其中所述至少三组多核苷酸中的每个组均包含多个多核苷酸;以及(e)将预定量的所述至少三组多核苷酸中的各个组混合以产生归一化对照。
本公开还提供了制备归一化对照组合物的方法,其中所述归一化对照包含使用本公开的方法合成的多核苷酸和包含使用本公开的方法分离和/或得到的分离的序列的多核苷酸两者。
本公开提供了使用归一化对照对样本中的至少一种靶核酸分子的表达水平进行定量的方法,所述方法包括:(a)将已知量的本公开的归一化对照与样本混合,(b)制备高通量测序文库,(c)对所述文库进行测序以产生读段集合,(d)将每个读段映射至样本或归一化对照,(e)确定由归一化对照中的多个核酸分子中的每个核酸分子产生的读段的数量,(f)计算(a)中与样本混合的归一化对照中的多个核酸分子中的每个核酸分子的起始浓度与(c)中产生的读段的数量之间的关系,(g)对读段与样本中的浓度之间的关系进行建模,以及(h)使用(g)中的模型,根据靶核酸分子产生的读段的数量计算样本中的至少一种靶核酸分子的初始浓度。
归一化对照组合物
本公开提供了归一化对照组合物(在本文中可互换地称为归一化对照,或(nc),其可用于对ngs应用中的样本输入进行定量。如本文所提供的,本公开的nc可包含至少两组多核苷酸,其中在所述至少两组中的每个组内的多核苷酸具有相同的长度。在一些实施方式中,将nc从头合成为多核苷酸。在一些实施方式中,nc是分离自或来源于样本,例如来自生物体的dna或rna样本,或载体。在一些实施方式中,nc包括从头合成的多核苷酸和分离自或来源于样本的多核苷酸两者。在一些实施方式中,nc包含至少三组多核苷酸,其中所述至少三组中的每个组内的多核苷酸具有相同的长度。
在一些实施方式中,nc包含至少3组不同长度的多核苷酸序列,并且每组多核苷酸以不同的浓度存在于归一化对照中。
在一些实施方式中,nc包含至少15组不同长度的多核苷酸序列,并且每组多核苷酸以不同的浓度存在于归一化对照中。
在一些实施方式中,nc包含15组不同长度的多核苷酸序列,并且每组多核苷酸以不同的浓度存在于归一化对照中。
nc中的多核苷酸长度
可以在整个浓度范围内改变归一化对照内的多核苷酸的长度,以允许片段化率或聚类的偏倚(例如有时在基于illumina的ngs方法的情况下看到的)。例如,归一化对照包括多组多核苷酸,并且每组多核苷酸具有不同的长度并且在归一化对照内以不同的浓度存在。
设想具有不同长度和长度的组合的各组多核苷酸在本公开的归一化对照的范围内。给定组内的所有单独多核苷酸均具有相同的长度。然而,在包含至少三个组的归一化对照组合物中,一个组内的多核苷酸的长度可以与归一化对照组合物中的任何其他组内的多核苷酸的长度相同或不同。
因此,在一些实施方式中,所述归一化对照中的所有多核苷酸的长度是相同的。在一些实施方式中,至少三个组中的每一个组内的多核苷酸具有与任何其他组中的多核苷酸不同的长度。
在一些实施方式中,至少三组多核苷酸的长度以线性序列或几何序列分布。
线性序列是数字序列,其中所述序列中的下一个数字每次相对于前一个数字增加或减少相同的量。示例性线性序列包括序列“1、2、3、4、5、6”。通常,线性序列由下式表示:
un=dxn+c
其中d是序列中连续项之间的第一个差异,n是序列中的项,并且c是常数。
几何序列是数字序列,其中第一项之后的每个项都可以通过将前一项乘以固定的非零数(称为“公共比率”)来找到。示例性的几何序列包括“2、4、8、16”(每个数字是前一个的2倍)。通常,几何序列由下式表示:
un=u1r-1
其中r是公共比率。
在一些实施方式中,例如在其中多核苷酸包含组分序列的那些实施方式中(如本文进一步描述的),所述组中的每个组中的多核苷酸的长度介于约15bp与约3000bp之间、介于约50bp与约3000bp之间、介于约100bp与约3000bp之间、介于约1000bp与约3000bp之间、介于约1200与约3000bp之间、介于约1500bp与约3000bp之间、介于约15bp与约50bp之间、介于约15bp与约100bp之间、介于约15bp与约150bp之间、介于约15bp与约200bp之间、介于约15bp与约300bp之间、介于约15bp与约400bp之间、介于约15与约500bp之间、介于约50bp与约1200bp之间、介于约100bp与约1200bp之间、介于约150bp与约1200bp或介于约150与约1100bp之间。在示例性实施方式中,各组多核苷酸中的多核苷酸的长度介于约15bp与约3000bp之间。在示例性实施方式中,各组多核苷酸中的多核苷酸的长度介于约500bp与约1500bp之间。在示例性实施方式中,各组多核苷酸中的多核苷酸的长度介于约100bp与约1200bp之间。在示例性实施方式中,各组多核苷酸中的多核苷酸的长度介于约150bp与约600bp之间。
在一些实施方式中,至少三组多核苷酸包含具有选自由以下组成的长度的多核苷酸:175bp、250bp和450bp;192bp、250bp和450bp;200bp、300bp和500bp;217bp、300bp和517bp;436bp、552bp和974bp;450bp、612bp和1034bp;510bp、626bp和1048bp;以及450bp、612bp和1034bp。
如本文所提供的,各组多核苷酸可以在体外合成,或者可以分离自或来源于已合成的核酸,或者可以分离自或来源于天然存在的核酸。预期在体外合成的多核苷酸可具有约3kb的最大长度,但是本领域技术人员将理解该长度可取决于当前的寡核苷酸合成技术。预期与使用合成方法制备的归一化对照相比,分离自或来源于已经合成的或天然存在的核酸的归一化对照多核苷酸可以包含更宽的长度范围,但是在此,所述长度范围也是取决于当前寡核苷酸合成技术。
在一些实施方式中,例如在归一化对照多核苷酸包含分离的序列(如本文进一步描述)的情况下,至少三个不同组的多核苷酸的长度介于约15bp与约500kb之间。在一些实施方式中,至少三个不同组的多核苷酸的长度介于约15bp与约50kb之间。在一些实施方式中,至少三个不同组的多核苷酸的长度介于约50bp与约50kb之间。在一些实施方式中,至少三个不同组的多核苷酸的长度介于约500bp与约50kb之间。在一些实施方式中,至少三个不同组的多核苷酸的长度介于约1000bp与约50kb之间。在一些实施方式中,至少三个不同组的多核苷酸中的每一组中的最长的是至少15kb的长度、至少20kb的长度、至少25kb的长度、至少30kb的长度、至少35kb的长度、或至少40k的长度,包括端点。
nc中的多核苷酸序列
设想本公开的各组多核苷酸包含具有相同序列的多核苷酸,或包含其中该组中的所有多核苷酸的序列不完全相同的多核苷酸。
在一些实施方式中,每个组内的多核苷酸由相同序列组成。
在一些实施方式中,每个组内的多核苷酸包含相同的序列。对于每个组中的所有多核苷酸可以相同的序列的非限制性示例包括衔接子序列、引物序列、索引序列、限制性位点、启动子序列、组分序列、分离的序列,或它们的组合。
在一些实施方式中,每个组内的多核苷酸包含在所有多核苷酸中相同的序列和在所有多核苷酸中不同的序列两者。例如,每个组内的所有多核苷酸可包含相同的衔接子和/或索引序列,但是它们的组分序列不同。在其他实施方式中,每个组内的所有多核苷酸可包含不同的衔接子和/或索引序列,但是共享相同的组分序列或分离的序列。
在一些实施方式中,每个组内的多核苷酸的序列是至少5%同一、至少10%同一、至少20%同一、至少30%同一、至少40%同一、至少50%同一的,是至少60%同一、至少70%同一、至少80%同一的,是至少90%同一、至少95%同一、至少97%同一、至少98%同一或至少99%同一的。
在一些实施方式中,每个组内的多核苷酸不包含相同的序列。
在一些实施方式中,一个或多个多核苷酸或每组多核苷酸包含多核苷酸的至少三个亚组,其中每个亚组内的多核苷酸包含相同的序列,并且每个亚组内的多核苷酸不包含与任何其他亚组相同的序列。
在本公开的归一化对照的一些实施方式中,至少三个组中的每个组内的每个多核苷酸包含相同的序列。
在一些实施方式中,至少三个组中的每个组内的多个多核苷酸的序列不包含与任何其他组中的多个多核苷酸的序列相同的序列。
在一些实施方式中,至少三个组中的每个组内的多个多核苷酸中的每一个多核苷酸包含不具有相同序列的多核苷酸。
在一些实施方式中,至少三组多核苷酸中的每一个组包含多核苷酸的至少三个亚组,并且多核苷酸的每个亚组包含具有相同序列的多个多核苷酸,并且每个亚组中的多个多核苷酸的序列不与该组中的其他亚组相同。
组分序列
在本公开的归一化对照的一些实施方式中,至少一组多核苷酸的序列包含被设计、选择或已知为具有某些特性(例如长度、序列或gc含量)的组分序列。这可以导致那组多核苷酸具有所表示的那些特性的经设计、选择或已知的范围。组分序列的性质可经设计或选择以匹配测序样本中的靶序列的特性。
如本文所用,术语“组分序列”是指在归一化对照中发现的多核苷酸的一部分或全部。归一化对照多核苷酸包含组分序列或由组分序列组成。在一些实施方式中,组分序列是计算机模拟设计的,并且不是分离自或来源于任何生物体的。
在一些实施方式中,组分序列包括随机序列。在一些实施方式中,将组分序列设计为模拟或共享测序样本中的靶序列的一种或多种序列特性。
在一些实施方式中,每组多核苷酸的序列包含组分序列。在一些实施方式中,每组多核苷酸的组分序列与任何其他组的组分序列不同。
在一些实施方式中,至少一组多核苷酸的至少一个亚组的序列包含组分序列。在一些实施方式中,至少一个组的至少三个亚组中的每一个亚组的序列包含组分序列。在一些实施方式中,每个组的至少三个亚组中的每一亚组的序列包含组分序列。在一些实施方式中,每个亚组的组分序列与任何其他亚组的组分序列不同。
例如,在具有三个组的归一化对照组合物中,每个组具有三个亚组,每个亚组具有不同的组分序列,所述组分序列没有一个是相同的。在该示例中,归一化对照组合物中的多核苷酸群体包含具有九种不同组分序列的多核苷酸。
在一些实施方式中,组分序列包含介于约15bp与约3000bp之间、介于约50bp与约3000bp之间、介于约100bp与约3000bp之间、介于约1000bp与约3000bp之间、介于约1200与约3000bp之间、介于约1500bp与约3000bp之间、介于约15bp与约50bp之间、介于约15bp与约100bp之间、介于约15bp与约150bp之间、介于约15bp与约200bp之间、介于约15bp与约300bp之间、介于约15bp与约400bp之间、介于约15与约500bp之间、介于约50bp与约1200bp之间、介于约100bp与约1200bp之间、介于约150bp与约1200bp或介于约150与约1100bp之间的序列。在一些实施方式中,组分序列包含介于约6bp与约3000bp之间的序列。在一些实施方式中,组分序列包含介于约150bp与约500bp之间的序列。
在本公开的方法的一些实施方式中,组分序列与测序样本中的靶序列不同。在一些实施方式中,组分序列具有与测序样本中的靶序列的小于或等于1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、30%、40%、50%、60%、70%、80%、85%、90%、95%、96%、97%、98%、99%或99.5%、99.8%或99.9%的同一性。在一些实施方式中,组分序列可与靶序列差异每20bp约1bp、每50bp约1bp、每150bp约1bp、每250bp约1bp、或每300bp约1bp。
在一些实施方式中,组分序列包含随机序列或由随机序列组成。在一些实施方式中,使用随机序列生成器来生成随机序列。在一些实施方式中,随机序列不映射到参考序列。在一些实施方式中,随机序列不映射到任何感兴趣的生物体(例如ncbi核苷酸数据库中的任何生物体,或将使用归一化对照来检测、定量或以其他方式分析的任何生物体)的基因组。在一些实施方式中,随机序列不由在被设计用于切割人dna的90kgrna阵列中的任何与crispr相关联的(crispr-associated,cas)指导rna(guiderna,grna)切割。在一些实施方式中,可以将随机序列设计为排除特定的序列特征。例如,随机序列可以排除诸如crispr/casgrna识别位点、限制性位点,转录因子结合位点或重复序列的特征。在一些实施方式中,可以将随机序列设计为模拟靶序列的特性。例如,随机序列可以被设计为具有与靶序列相同的gc含量或相同的特定序列基序(例如三核苷酸重复)频率。
分离的序列
在本公开的归一化对照的一些实施方式中,归一化对照多核苷酸包含分离的序列。如本文所用,“分离的序列”是一种类型的组分序列。术语“分离的序列”是指已经分离自或来源于生物体或其他先前存在的序列的序列。分离的序列可包含基因组、线粒体或叶绿体dna序列。分离的序列可以包含rna序列,例如信使rna(messengerrna,mrna)、转移rna(transferrna,trna)、非编码rna(non-codingrna,ncrna)或微型rna。分离的序列可以分离自生物体或预先存在的序列,并且包含预先存在的序列的一个或多个片段。替代地或另外,可以对分离的序列进行一种或多种转化,以从预先存在的序列中得到它们,所述一种或多种转化为例如序列改组、序列连接、反转核苷酸的顺序、或嵌套在其他序列中。
在一些实施方式中,至少一组多核苷酸的序列包含分离的序列。在一些实施方式中,每组多核苷酸的序列包含分离的序列。
在一些实施方式中,至少一组多核苷酸的至少一个亚组的序列包含分离的序列。在一些实施方式中,至少一个组的每个亚组的序列包含分离的序列。在一些实施方式中,每个组的每个亚组的序列包含分离的序列。在一些实施方式中,每个亚组的分离序列与任何其他亚组的分离序列不同。
在一些实施方式中,分离的序列的长度介于约15bp与约500kb之间。在一些实施方式中,分离的序列的长度介于约15bp与约100kb之间。在一些实施方式中,分离的序列的长度介于约15bp与约50kb之间。在一些实施方式中,分离的序列的长度介于约50bp与约50kb之间。在一些实施方式中,分离的序列的长度介于约500bp与约50kb之间。在一些实施方式中,分离的序列的长度介于约1000bp与约50kb之间。
在一些实施方式中,分离的序列介于约6bp与约200,000bp之间、介于约15bp与约50,000bp之间、介于约500bp与约1500bp之间、介于约100bp与约1200bp之间,或介于约150bp与约600bp之间。
在一些实施方式中,分离的序列为至少约50bp、至少约100bp、至少约150bp、至少约200bp、至少约250bp、至少约300bp、至少约350bp、至少约400bp、至少约450bp、至少约500bp、至少约550bp、至少约600bp、至少约650bp、至少约600bp、至少约750bp、至少约800bp、至少约850bp、至少约900bp、至少约950bp、至少约1000bp、至少约1250bp、至少约1500bp、至少约2000bp、至少约2500bp或至少约3000bp。
在一些实施方式中,分离的序列为约100bp、约200bp、约300bp、约400bp、约500bp、约600bp、约700bp、约800bp、约900bp、约1000bp、约1100bp、约1200bp、约1300bp、约1400bp或约1500bp。
在一些实施方式中,分离的序列是分离的或或来源于病毒、细菌、真菌或真核寄生虫。在一些实施方式中,病毒是t4噬菌体(t4)或巨细胞病毒(cmv)。在一些实施方式中,分离的序列分离自克隆载体(例如质粒)。
在一些实施方式中,从其纯化归一化对照的分离的序列的核酸样本是克隆载体。在一些实施方式中,克隆载体是细菌组分染色体(bacterialcomponentchromosome,bac)、酵母人工染色体(yeastartificialchromosome,yac)、粘粒、福斯质粒或质粒。在一些实施方式中,从其纯化分离的归一化对照的核酸样本是质粒。在一些实施方式中,将归一化对照序列从感兴趣的物种(例如病毒、细菌或真核寄生虫)克隆到克隆载体中,并使用本公开的方法从克隆载体中纯化归一化对照。
在一些实施方式中,分离的序列与测序样本中的靶序列不同。在一些实施方式中,归一化对照中的任何多核苷酸的序列具有与测序样本中的靶序列小于或等于1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、30%、40%、50%、60%、70%、80%、85%、90%、95%、96%、97%、98%或99%的同一性。
在一些实施方式中,分离的序列分离自或来源于多个参考序列。“参考序列”是指与本领域中已知的靶序列相同或相似的一个或多个序列。参考序列可以指单个靶序列,例如靶生物体的基因或基因组。
作为另一个示例,“参考序列”可以指来自多种不同生物体的多种序列。所述生物体可以是病毒、细菌、真菌或单细胞真核生物,例如寄生或致病性真核生物,或它们的组合。参考序列可以是基因组dna序列、cdna序列或它们的组合。
在一些实施方式中,归一化对照包含分离自或来源于单个参考序列的多个分离的序列。
在一些实施方式中,归一化对照包括多个分离的序列,所述多个分离的序列分离自或来源于多个参考序列,例如来自多种不同的生物体。在一些实施方式中,多个参考序列包括来自通常被认为是非宿主生物体的生物体的多个序列,所述生物体为例如病毒、细菌或真菌(例如,当宿主是哺乳动物、植物或多细胞真核生物时)。多个参考序列可用于对本文所述的归一化对照中的靶生物体组的群体水平特征进行建模。在测序样本中可以存在任何一组靶生物体,并且它们的存在和滴度可以使用本文所述的方法进行测量。在其中测序样本中的靶序列或靶生物体在测序之前未知的那些实施方式中,使用对多种生物体进行建模的归一化对照可以提高鉴定和滴度测量的准确性。
在一些实施方式中,多个分离的序列是分离自或来源于至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少20个、至少50个、至少100个、至少200个、至少300个、至少400个、至少500个、至少600个、至少700个、至少800个、至少900个、至少1,000个、至少1,200个、至少1,300个、至少1,400个、至少1,500个、至少1,600个、至少1,700个、至少1,800个、至少1,900个、至少2,000个、至少2,200个、至少2,400个、至少2,600个、至少2,800个、至少3,000个、至少4,000个、至少5,000个、至少6,000个、至少7,000个、至少8,000个、至少9,000个、至少10,000个参考序列、至少11,000个参考序列、或至少12,000个参考序列。在一些实施方式中,多个分离的序列是分离自或来源于至少10个参考序列。在一些实施方式中,多个分离的序列是分离自或来源于至少100个参考序列。在一些实施方式中,多个分离的序列是分离自或来源于至少500个参考序列。来源于在一些实施方式中,多个分离的序列是分离自或来源于至少1000个参考序列。来源于在一些实施方式中,多个分离的序列是分离自或来源于至少1,200个参考序列。来源于在一些实施方式中,多个分离的序列是分离自或来源于至少1,500个参考序列。来源于在一些实施方式中,多个分离的序列是分离自或来源于至少2,000个参考序列。在一些实施方式中,每个参考序列来自不同生物体或同一生物体的不同品系或亚种。在一些实施方式中,多个参考序列是来自不同生物体或同一生物体的不同品系或亚种的基因组dna序列。
在一些实施方式中,多个参考序列包括来自多种生物体的参考序列。在一些实施方式中,多个参考序列包含来自至少2种、至少10种、至少20种、至少50种、至少100种、至少200种、至少300种、至少400种、至少500种、至少600种、至少700种、至少800种、至少900种、至少1,000种、至少1,200种、至少1,300种、至少1,400种、至少1,500种、至少1,600种、至少1,700种、至少1,800种、至少1,900种、至少2,000种、至少2,200种、至少2,400种、至少2,600种、至少2,800种、至少3,000种、至少4,000种、至少5,000种、至少6,000种、至少7,000种、至少8,000种、至少9,000种、或至少10,000种生物体的序列。在一些实施方式中,多个参考序列包含来自约1,000种生物体、来自约1,200种生物体、来自约1,400种生物体、来自约1,500种生物体、来自约1,600种生物体、来自约1,700种生物体、来自约1,800种生物体、来自约2,000种生物体、来自约2,200种生物体、来自约2,400种生物体、来自约2,500种生物体、来自约3,000种生物体、来自约5,000种生物体、来自约7,000种生物体、或来自约10,000种生物体的序列。
在一些实施方式中,多个分离的序列包含分离自或来源于参考序列的片段的序列。在一些实施方式中,参考序列片段是参考序列的介于约10-100个之间、介于约10-80个之间、介于约10-70个之间、介于约10-60个之间、介于约10-50个之间、介于约10-40个之间、介于约10-30个之间、介于约10-20个之间、介于约20-100个之间、介于约20-80个之间、介于约20-60个之间、介于约20-50个之间、介于约20-40个之间、介于约20-30个之间、介于约10-35个之间、介于约20-35个之间、或介于约20-25个之间的连续核苷酸。在一些实施方式中,所述参考序列片段是参考序列的介于约15-60个之间、介于约20-40个之间、介于约20-30个之间、介于约15-32个之间、介于20-32个个之间或介于约25-35个之间的连续核苷酸。在一些实施方式中,参考序列片段包含参考序列的20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、31个、32个、33个、34个、35个、36个、37个、38个、39个、40个、41个、42个、43个、44个、45个、46个、47个、48个、49个、50个、51个、52个、53个、54个、55个、56个、57个、58个、59个、60个、61个、62个、63个、64个或65个连续碱基对。在一些实施方式中,参考序列片段包含参考序列的25个、26个、27个、28个、29个、30个、31个、32个、33个、34个或35个连续bp。在一些实施方式中,参考序列片段包含参考序列的31个或32个连续bp。
在一些实施方式中,参考序列片段小于测序读段的平均长度的一半。例如,如果ngs测序方法产生的平均读段均为120bp,则参考序列片段小于60bp。不同的ngs平台可产生不同长度的测序读段。例如,hiseq和hiseqx的平均读段长度为约300bp,而miseq平台的平均读段长度介于约300bp与600bp之间。在一些实施方式中,平均ngs读段长度为约100bp、约150bp、约200bp、约250bp、约300bp、约350bp、约400bp、约450bp、约500bp、约550bp、约600bp、约650bp、约700bp、约750bp、约800bp、约850bp、约900bp、约950bp、约1000bp、约1100bp、约1200bp、约1300、约1400bp、约1500bp、约1600bp、约1700bp、约1800bp、约1900bp或约2000bp。
在一些实施方式中,多个分离的序列包含已经改组的参考序列的片段。改组序列的方法在本领域中是已知的,并且包括随机改组和cantor改组。各种程序可用于改组核苷酸序列,并且将是本领域技术人员已知的(参见例如,www.bioinformatics.org/sms2/shuffle_dna.html)。如本文中所预期的,经改组的参考序列片段保留了参考序列的一种或多种特征,而与此同时与样本中的靶序列不同,并且因此期望通过例如blast的方法与靶序列相区分。
在一些实施方式中,多个分离的序列包含已连接的参考序列片段。在一些实施方式中,将参考序列片段改组并连接,例如以产生具有期望的一个或多个长度的分离的序列。在一些实施方式中,分离的序列包含至少第一参考序列和第二参考序列的至少两个片段,所述至少两个片段已被改组和连接。在一些实施方式中,第一参考序列和第二参考序列在生物体的基因组中不相邻。在一些实施方式中,第一参考序列和第二参考序列来自不同的生物体。分离的序列可以包含至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个参考序列片段,所述参考序列片段已经被改组并连接,其中所述参考序列片段中的任何一个参考序列片段都可能是来自相同参考序列或参考生物体,或来自不同参考序列或参考生物体的不相邻片段。
在一些实施方式中,分离的序列是通过以下过程而来源于参考序列的:(a)通过参考序列片段的分布来表示至少一个参考序列;(b)划分成箱;(c)从所述箱的至少一个子集中选择多个参考序列片段;以及(d)对参考序列片段进行改组并任选地连接以生成分离的序列。
在一些实施方式中,通过以下方式生成归一化对照的分离的序列:(a)生成在至少一个参考序列上使用滑动窗生成的重叠参考序列片段的分布;(b)将所述分布划分成箱;(c)从所述箱的至少一个子集中选择多个参考序列片段;以及(d)连接所述参考片段序列,从而生成分离的序列。
在一些实施方式中,滑动窗包含1个碱基对(bp)、2bp、3bp、4bp、5bp、6bp、7bp、8bp、9bp、10bp、11bp、12bp、13bp、14bp、15bp、16bp、17bp、18bp、19bp或20bp的滑动窗。滑动窗是长度为n的窗,其可沿参考序列滑动以生成参考序列片段,所述参考序列片段会偏移所述窗的长度达例如1bp、2bp、3bp、4bp、5bp或更多。
该分布可以是以下各项中任一项的分布:(1)gc含量百分比、(2)熵、(3)复杂性、(4)电子-离子相互作用电势(eiip)、(5)长度,或它们的组合。为了生成分布,将参考序列分解为具有一个或多个指定大小的片段(有时在本文中称为kmer),并将参考序列片段的数量针对给定的参数(例如gc含量百分比、eiip、长度或熵)进行分箱。每个箱代表参数分布的百分比,例如1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、15%、20%、或25%。
分布可以划分成任意数量的箱。例如,可以将分布划分成2、3、4、5、6、7、8、9、10、20、50、100或1000个箱。在一些实施方式中,每个箱代表参数(例如gc含量百分比、熵、复杂性、长度或eiip)的分布的0.1%、0.5%、1%、2%、5%、10%、15%或20%。
在一些实施方式中,使用选自划分成5个箱的分布的至少3个箱中的一个箱的参考序列片段,来生成归一化对照的每个组内的多核苷酸。在一些实施方式中,通过使用选自分布中的至少1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个或20个箱的参考序列片段来生成归一化对照中的每个组内的多核苷酸。在一些实施方式中,将分布划分成10个箱,并且参考序列片段选自来自gc含量百分比分布的第10、第30、第50、第70、第80、和第90百分位箱。在一些实施方式中,通过使用选自分布中每个箱的参考序列片段来生成归一化对照的每个组内的多核苷酸。
在一些实施方式中,一个或多个参考序列片段选自分布(例如gc含量百分比分布)的一个箱。在一些实施方式中,从分布中的每个箱中选择一个或多个参考序列片段。在一些实施方式中,从分布的箱的子集中选择一个或多个参考序列片段。在一些实施方式中,将从相同箱中选择的参考序列片段连接起来以形成具有期望长度的分离的序列。这允许归一化对照中的分离的序列通过对选定的箱进行子取样来对整个分布范围内的各种参数(例如gc含量、eiip、长度和熵)的分布进行建模,而无需以参考序列与归一化对照之间的1:1对应关系,使用高得离谱的数量的归一化对照寡核苷酸来对整个分布参考序列进行建模。
在一些实施方式中,每个组内的多核苷酸具有以下项中的至少三者:与来自所述参考序列分布中的对应箱的参考序列片段(1)相似的gc含量百分比、(2)相似的熵、(3)相似的eiip、(4)相似的长度、或(5)相似的复杂性。
相似的gc含量百分比、eiip、长度、复杂性或熵可以指参考序列和归一化对照的平均gc含量、eiip、长度、复杂性或熵的比较。例如,如果参考序列的平均gc含量、eiip、长度、复杂性或熵在归一化对照的平均gc含量、eiip、长度、复杂性或熵的0.1、0.5、1、2、3、4、5、6、7、8、9、10、11、12、13、14或15个百分点内,则归一化对照和参考序列可具有类似的gc含量百分比、eiip、长度、复杂性或熵。替代地或另外,类似的gc含量、eiip、长度、复杂性或熵可以指参考序列和归一化对照序列中的一些或全部序列上的gc含量、eiip、长度、复杂性或熵的分布。确定相似程度的方法将是本领域中是已知的,包括但不限于,kolmogorov-smirnov检验、z检验、q-q取向和方差分析(analysisofvariance,anova)。普通技术人员将能够基于所讨论的分布的特征(例如,正态、泊松、高斯或偏斜)选择适当的检验。
在一些实施方式中,多个分离的序列包含已连接的参考序列片段。在一些实施方式中,参考序列片段被连接而不被改组。如果经连接以形成分离的序列的参考序列片段在参考序列中不相邻,或者来自不同参考序列,则在不相邻序列之间或来自不同来源的序列之间存在“接头”应允许在ngs后通过blast比对鉴定分离的序列。接头的存在允许归一化对照中的分离的序列与样本中的靶序列区分开。不希望受理论的束缚,预期对于这种方法,介于约15bp与约50bp之间,介于约20bp与约35bp之间的序列片段可为优选的,因为太小的序列,例如低于比对器的种子长度的序列,架构可能不足以独特地产生稳健的比对,而太大的序列将不会具有通过单独ngs读段捕获的接头。
在一些实施方式中,分离的序列包含连接的至少一个参考序列的至少两个片段,并且所述至少两个片段在至少一个参考序列中不相邻。即,至少两个片段分开至少1bp、至少2bp、至少3bp、至少4bp、至少5bp、或任何数量的bp。
在一些实施方式中,分离的序列包含至少第一参考序列的至少第一片段和至少第二参考序列的至少第二片段,其中所述至少第一和第二片段是连接的,并且其中所述第一和第二参考序列不是相同的参考序列。例如,第一片段来自人腺病毒,并且第二片段来自白色念珠菌(candidaalbicans)。作为进一步的示例,分离的序列可包含嵌套的参考序列片段:例如,来自第二参考生物体的参考序列片段内的来自第一参考生物体的参考序列片段,任选地与来自另一种不同生物体的参考序列片段连接。分离的序列可包含至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个参考序列片段,所述参考序列片段已经被连接,其中所述参考序列片段中的任何一个参考序列片段都可能是来自相同参考序列或参考生物体,或来自不同参考序列或参考生物体的不相邻片段。
在一些实施方式中,分离的序列(例如,改组或连接的分离的序列)不被设计用于切割人dna的90kgrna阵列中的任何crispr相关(cas)指导rna(grna)切割。在一些实施方式中,分离的序列可以被设计为排除特定的序列特征。例如,可以选择不包含诸如crispr/casgrna识别位点、限制性位点、转录因子结合位点或重复序列的特征的改组或连接的分离的序列。
序列特性
在一些实施方式中,至少三组多核苷酸中的每个组的序列具有与测序样本中的靶序列共有的至少一种特性。在一些实施方式中,将组分序列设计为具有与测序样本中的靶序列共有的至少一种特性。在一些实施方式中,分离的序列是从已经选择的生物体或克隆载体中的序列中分离的,因为已知该分离的序列具有与测序样本中的靶序列共有的至少一种特性。
在一些实施方式中,至少一种特性包括转座元件序列、病毒序列、细菌序列、真菌序列、真核寄生虫序列、或一个或多个人类基因序列的序列特性。
在一些实施方式中,转座元件的特性包括转座元件序列。转座元件是可以改变基因组中的位置的dna序列。转座元件(转座子)通常由一对反向末端重复序列(invertedterminalrepeat,itr)组成,该itr位于编码转座酶的开放阅读框的侧翼。因此,转座元件的示例性特性包括itr序列、转座酶序列或转座子的任何序列。转座子的任何序列或转座子的序列特性可被并入本公开的组分序列中。本公开的分离的序列可包含转座子的任何序列或转座子的序列特性。
在一些实施方式中,病毒序列的特性包括gc含量百分比、重复序列元件、反向末端重复(itr)、内部核糖体进入位点(ires)、蛋白质编码序列、转录后调控元件、转录调控元件、启动子序列、顺式作用rna元件、rna结构元件、基因组包装信号、5'非翻译区(5'utr)序列、3'非翻译区(3'utr)序列,或它们的组合。可以将病毒的任何序列或病毒的序列特性并入本公开的组分序列中。
在一些实施方式中,细菌序列的特性包括gc含量百分比、重复序列、微型rna结合位点、内部核糖体进入位点(ires)、蛋白质编码序列、转录调控元件、启动子序列、5'utr序列、3'utr序列,或它们的组合。在一些实施方式中,重复序列元件包含polya基序、polyt基序、polyg基序、polyc基序、二核苷酸基序、三核苷酸基序、四核苷酸基序、五核苷酸基序、六核苷酸基序、七核苷酸基序、八核苷酸基序、非核苷酸基序、散布的重复序列元件、核糖体rna序列、转移rna(trna)序列、末端反向重复序列(tir)、非自主微型反向重复转座元件(non-autonomousminiatureinvertedrepeat,mite)、成簇规律间隔短回文重复序列(clusteredregularlyinterspacedshortpalindromicrepeat,crispr),或它们的组合。polya基序、polyt基序、polyg基序和polyc基序是一系列a、t、g和c(例如,4-50个a、t、g或c)。细菌的任何序列或细菌的序列特性都可以并入本公开的组分序列中。本公开的分离的序列可以包含细菌的序列或细菌的序列特性。
在一些实施方式中,真菌序列的特性包括gc含量百分比、重复序列元件、微型rna结合位点、内部核糖体进入位点(ires)、蛋白质编码序列、转录调控元件、启动子序列、5'utr序列、3'utr序列、着丝粒序列、端粒序列、子端粒序列,线粒体序列,或它们的组合。可以将真菌的任何序列或真菌的序列特性并入本公开的组分序列中。本公开的分离的序列可以包含真菌的序列或真菌的序列特性。
在一些实施方式中,一个或多个人类基因的特性包括gc含量百分比、重复序列、蛋白质编码序列、内含子序列、5'utr序列、3'utr序列、转录调控元件序列、启动子序列、微型rna结合序列,或它们的组合。在示例性实施方式中,一个或多个人类基因的特性是编码由一个家族中的多个蛋白质共享的保守蛋白质结构域的序列。蛋白质结构域的非限制性示例是dna结合结构域,例如锌指dna结合结构域或同源结构域。在示例性实施方式中,一个或多个人类基因的特性是由同源微型rna调控的所有基因共享的微型rna结合位点。可以将一个或多个人类基因的任何序列或一个或多个人类基因的序列特性并入本公开的组分序列中。本公开的分离的序列可以包含一个或多个人类基因的序列或一个或多个人类基因的序列特性。
在一些实施方式中,真核寄生虫序列的特性包括gc含量百分比、重复序列、微型rna结合位点、蛋白质编码序列、转录调控元件、启动子序列、5'utr序列、3'utr序列、着丝粒序列、端粒序列、子端粒序列、线粒体序列,或它们的组合。真核寄生虫的任何序列或序列特性都可以并入本公开的组分序列中。本公开的分离的序列可以包含真核寄生虫的序列或真核寄生虫的序列特性。
在一些实施方式中,归一化对照序列和靶序列共享的至少一种特性包括熵。熵是核苷酸序列的信息含量和复杂性的量度。计算熵的方法对于本领域普通技术人员而言将是已知的(参见例如,bioinformatics,第27卷,2011,第1061-1067页)。
在一些实施方式中,归一化对照序列和靶序列共享的至少一种特性包括复杂性。复杂性(用n表示)是在给定区段的多核苷酸或基因组的组件中的独特或非重复核苷酸的碱基对的数量。如果多核苷酸中的一些多核苷酸被重复,则这与序列的长度(l)不同。
在一些实施方式中,归一化对照序列和靶序列共享的至少一种特性包括gc含量百分比。
在本公开的归一化对照的一些实施方式中,多核苷酸的一个或多个组或亚组的gc含量与测序样本中的靶序列相同。在一些实施方式中,该组或亚组中的整个多核苷酸的gc含量与靶序列相同。在一些实施方式中,组分序列和/或分离的序列的gc含量与靶序列相同。已知gc含量会影响ngs期间的读段计数(gc偏差)。具有低或高gc含量的序列趋向于在ngs期间产生的读段数量不足。因此,在nc组合物中模拟靶序列的gc含量增加了nc对ngs期间的靶序列的行为进行建模的能力。
在一些实施方式中,一个组内的多核苷酸具有相同的gc含量百分比。在一些实施方式中,一个组内的多核苷酸具有与每个其他组内的多核苷酸相同的gc含量百分比。在一些实施方式中,一个组内的多核苷酸不具有与任何其他组内的多核苷酸相同的gc含量百分比。
在一些实施方式中,每个组内的多核苷酸的gc含量百分比介于40%与60%之间,包括端点。在一些实施方式中,每个组内的多核苷酸的gc含量百分比介于43%与56%之间,包括端点。
在一些实施方式中,归一化对照序列和靶序列共享的至少一种特性包括电子-离子干扰电势(eiip)。eiip可以定义为核苷酸序列中的核苷酸的离域电子的平均能量。通过将每个核苷酸替换为对应的eiip值,可以将核苷酸序列转换为数字eiip序列。例如,a=0.1260,c=0.1340,t=0.1335,g=0.0806。在一些实施方式中,eiip值可以在整个或部分序列上求和,例如在分离的序列或参考序列片段上求和。
在一些实施方式中,归一化对照序列和靶序列共享的至少一种特性包括gc含量、eiip和熵。gc含量、eiip和熵均取决于序列,因此共同变化。在一些实施方式中,归一化对照与靶序列共享这些参数中的所有三个参数。
可以通过本领域中已知的多种统计方法来确定归一化对照序列与靶序列或参考序列之间的参数(例如gc含量百分比、eiip、熵或复杂性)的相似程度。这些统计方法包括但不限于方差分析(anova)、t检验(例如学生t检验)、kolmogorovsmirnov(ks)检验和q-q曲线。在一些实施方式中,归一化对照包含经修饰的核苷酸。设想在本公开的范围内的任何类型的核苷酸修饰。下文描述本公开的核苷酸修饰的示例性但非限制性示例。
由本公开的方法使用的核苷酸修饰可以在任何核苷酸(例如腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶或尿嘧啶)上发生。这些核苷酸修饰可发生在脱氧核糖核酸(dna)或核糖核酸(rna)上。这些核苷酸修饰可以发生在双链或单链dna分子上,或发生在双链或单链rna分子上。
在一些实施方式中,核苷酸修饰包括腺嘌呤修饰或胞嘧啶修饰。
在一些实施方式中,腺嘌呤修饰包括腺嘌呤甲基化。在一些实施方式中,腺嘌呤甲基化包括n6-甲基腺嘌呤(6ma)。n6-甲基腺嘌呤(6ma)存在于原核基因组和真核基因组两者中。
在一些实施方式中,腺嘌呤甲基化包括ecoki甲基化。ecoki甲基化是由ecoki甲基化酶进行的一种类型的dna核苷酸修饰。ecoki甲基化酶修饰序列aac(n6)gtgc和gcac(n6)gtt中的腺嘌呤残基。在原核生物中发现了ecoki甲基化酶和ecoki甲基化。
在一些实施方式中,腺嘌呤修饰包括在n6处通过甘氨酸修饰的腺嘌呤(momylation)。甲基化改变n6-(1-乙酰氨基)-腺嘌呤的腺嘌呤。甲基化作用发生在病毒例如噬菌体中。
在一些实施方式中,修饰包括胞嘧啶修饰。在一些实施方式中,基因组中胞嘧啶修饰的丰度和类型基于物种而变化。在一些实施方式中,基因组中胞嘧啶修饰的位置(在例如特定的限制性内切酶识别位点内)基于物种而变化。
在一些实施方式中,胞嘧啶修饰包括5-甲基胞嘧啶(5mc)、5-羟基甲基胞嘧啶(5hmc)、5-甲酰基胞嘧啶(5fc)、5-羧基胞嘧啶(5cac)、5-葡萄糖基羟甲基胞嘧啶(5ghmc)或3-甲基胞嘧啶(3mc)。
在一些实施方式中,胞嘧啶修饰包括胞嘧啶甲酰化。在一些实施方式中,胞嘧啶甲基化包括5-甲基胞嘧啶(5mc)或n4-甲基胞嘧啶(4mc)。在一些实施方式中,在细菌中发现了4mc胞嘧啶甲基化。在一些实施方式中,胞嘧啶甲基化包括dcm甲基化。在一些实施方式中,胞嘧啶甲基化包括dnmt1甲基化,dnmt3a甲基化或dnmt3b甲基化。
在一些实施方式中,胞嘧啶甲基化包括cpg甲基化、cpa甲基化、cpt甲基化、cpc甲基化,或它们的组合。在一些实施方式中,胞嘧啶甲基化包括cpg甲基化。
在一些实施方式中,胞嘧啶修饰包括5-羟甲基胞嘧啶(5hmc)、5-甲酰基胞嘧啶(5fc)、5-羧胞嘧啶(5cac)、5-葡糖基羟甲基胞嘧啶、或3-甲基胞嘧啶。
靶生物体
本公开提供了用于确定样本中的一种或多种靶生物体的滴度的组合物和方法。靶生物体是包含一种或多种本文所述靶序列的生物体。
在一些实施方式中,样本包含宿主生物体和一种或多种非宿主生物体的混合物,并且靶生物体是一种或多种非宿主生物体。在一些实施方式中,宿主生物体是靶生物体。
在一些实施方式中,非宿主靶生物体是病原体。在一些实施方式中,靶生物体是病毒、细菌、真菌或真核病原体。例如,在一些实施方式中,靶生物体是人类临床样本中的病毒病原体,并且靶序列是该病毒病原体的序列。
在一些实施方式中,靶生物体是共生体或共生生物体。
在一些实施方式中,靶生物体是宏基因组样本中的物种混合物中的物种。
靶序列
在一些实施方式中,将nc设计为考虑影响测序样本中靶序列向ngs后的测序读段的转化的可能变量。例如,nc序列可经设计以匹配靶序列的核酸的gc含量,所述靶序列的在起始样本中的丰度是使用本公开的归一化对照和方法确定的。
在本公开的归一化对照的一些实施方式中,组分序列和/或分离的序列具有与测序样本中的靶序列共有的至少一种特性。在靶序列与组分序列之间共享的共同特性使得nc的组分序列能够在样本和/或文库制备和测序期间模拟靶序列。这允许nc对从样本中的靶序列的初始浓度至ngs之后靶序列的多个读段的转化进行建模。
在一些实施方式中,归一化对照中的多核苷酸的至少一个亚组的序列与测序样本中的靶序列共享至少一种序列特性。在一些实施方式中,至少一种序列特性包括转座元件序列、病毒序列、细菌序列、真菌序列、真核寄生虫序列,或一种或多种人类基因序列的序列特性。
预期靶序列的所有序列特性都在本公开的归一化对照组合物的范围内。在一些实施方式中,至少一种特性包括转座元件序列、病毒序列、细菌序列、真菌序列、真核寄生虫序列、或一个或多个人类基因序列的序列特性。在一些实施方式中,至少一种特性包括gc含量、熵、复杂性、长度、eiip,或它们的组合。
在一些实施方式中,测序样本包含宿主核酸和非宿主核酸的混合物。在一些实施方式中,非宿主包含一个或多个靶序列。
在一些实施方式中,宿主是真核生物。在一些实施方式中,宿主是昆虫、动物或植物。在一些实施方式中,动物是人。
在一些实施方式中,宿主是任何哺乳动物生物体。在特定的实施方式中,哺乳动物是人。在其他实施方式中,哺乳动物是牲畜动物,例如马、绵羊、牛、猪或驴。在其他实施方式中,哺乳动物生物体是家养宠物,例如猫、狗、沙鼠、小鼠或大鼠。在其他实施方式中,哺乳动物是一种类型的猴子。
在一些实施方式中,宿主是任何鸟类或禽类生物体。禽类生物体包括但不限于鸡、火鸡、鸭和鹅。
在一些实施方式中,宿主是昆虫。昆虫包括但不限于蜜蜂、独居蜂、蚂蚁、苍蝇、黄蜂和蚊子。
在一些实施方式中,宿主是植物。在一些特定的实施方式中,植物是稻、玉米、小麦、玫瑰、葡萄、咖啡、水果、番茄、马铃薯或棉花。
在一些实施方式中,非宿主包含共生体、共生生物体、寄生虫或病原体。在一些实施方式中,非宿主包含一种或多种物种的共生体、共生生物体、寄生虫或病原体。
在一些实施方式中,非宿主是病原体。在一些实施方式中,非宿主是病毒物种、细菌物种、真菌物种、或真核寄生虫物种。在一些实施方式中,非宿主是藻类物种。
在一些实施方式中,真核寄生虫是哺乳动物寄生虫。在一些实施方式中,该寄生虫是蠕虫。在其他实施方式中,该寄生虫是引起疟疾的寄生虫。在其他实施方式中,该寄生虫是引起利什曼病的寄生虫。在其他实施方式中,该寄生虫是变形虫。
在一些实施方式中,非宿主是细菌物种。在特定的实施方式中,细菌是引起结核的细菌。
在一些实施方式中,靶序列是测序样本中的非宿主序列。
在一些实施方式中,靶序列包含参考序列。在一些实施方式中,靶序列包含多个参考序列。在一些实施方式中,靶序列包括至少2个、至少10个、至少20个、至少50,least100个、至少200个、至少300个、至少400个、至少500个、至少600个、至少700个、至少800个、至少900个、至少1,000个、至少1,200个、至少1,300个、至少1,400个、至少1,500个、至少1,600个、至少1,700个、至少1,800个、至少1,900个、至少2,000个、至少2,200个、至少2,400个、至少2,600个、至少2,800个、至少3,000个、至少4,000个、至少5,000个、至少6,000个、至少7,000个、至少8,000个、至少9,000个或至少10,000个参考序列。在一些实施方式中,每个参考序列来自不同的生物体。在一些实施方式中,多个参考序列是来自不同生物体的基因组dna或cdna序列。在一些实施方式中,靶序列包含来自至少2种、至少10种、至少20种、至少50,least100种、至少200种、至少300种、至少400种、至少500种、至少600种、至少700种、至少800种、至少900种、至少1,000种、至少1,200种、至少1,300种、至少1,400种、至少1,500种、至少1,600种、至少1,700种、至少1,800种、至少1,900种、至少2,000种、至少2,200种、至少2,400种、至少2,600种、至少2,800种、至少3,000种、至少4,000种、至少5,000种、至少6,000种、至少7,000种、至少8,000种、至少9,000种或至少10,000种生物体的多个参考序列。
在一些实施方式中,靶序列包含非宿主序列,并且非宿主是病毒物种、细菌物种、真菌物种或真核寄生虫物种。
在一些实施方式中,非宿主包含共生体。共生体,有时称为互惠或共生生物体,是与另一种生物体有关系的生物体,所述关系可为双方带来互利。
在一些实施方式中,非宿主包含共生生物体。在共生生物体相互作用中,一种生物体(例如,非宿主)受益,而另一种(例如,宿主)则不受影响。共生生物体或互惠生物体可能取决于其宿主的食物栖息地(foodshelter)、支撑、运输或它们的组合。宿主可以从相互作用中获得多种益处,包括但不限于防止感染、改善消化和免疫调节。互惠和/或共生关系的非限制性示例是人类宿主与肠道内居住的微生物群体(肠道微生物群、或微生物群系)的互惠和/或共生关系。肠道菌群帮助维持肠道粘膜屏障,提供营养物质(例如维生素),预防病原体,并有助于维持适当的免疫功能。
在一些实施方式中,非宿主包括多个物种,或来自相同物种的不同生物体。在一些实施方式中,非宿主包含至少5种、至少10种、至少50种、至少100种、至少200种、至少500种、至少1000种、至少1,500种、或至少2,000种物种,或来自相同物种的不同生物体,或它们的组合。在一些实施方式中,非宿主包含至少2,000种物种,例如至少2,000种肠道微生物群系物种或微生物群系生物体。
在一些实施方式中,靶序列是与致病性相关的非宿主序列。与致病性相关的示例性非宿主序列包括但不限于与真核寄生虫相关的抗生素抗性基因,毒力因子或标志物致病性。
毒力因子是在感染宿主时有助于微生物如细菌,病毒或真菌致病性的基因或一组基因。毒力是指微生物对宿主造成的损害程度,更大的毒力导致更大的损害。
示例性但非限制性的毒力因子包括有助于宿主中利基定殖的基因(例如,非宿主与宿主细胞的附着),免疫逃避(逃避宿主免疫应答),免疫抑制(抑制肝细胞毒性)。宿主的非宿主免疫应答),(细胞内非宿主的)细胞进入和退出,以及从宿主获得营养。用于辅助和促进宿主定殖的示例性毒力因子包括但不限于粘附素,侵袭素和抗吞噬因子。
示例性毒力因子包括增加对宿主损害的因子,包括但不限于调节宿主gtp酶的毒素,破坏性酶和蛋白质。示例性毒素包括内毒素和外毒素。内毒素(脂多糖或lps)是一种细菌细胞壁成分,能够触发宿主的炎症。外毒素是由某些细菌主动分泌的毒素,对宿主具有广泛的作用,包括但不限于抑制特定的生化途径。示例性外毒素包括但不限于破伤风梭菌分泌的破伤风毒素,肉毒梭菌分泌的肉毒杆菌毒素,以及大肠杆菌,产气荚膜梭菌,炭疽芽孢杆菌和艰难梭菌分泌的毒素。在一些实施方式中,真菌分泌内毒素(称为霉菌毒素)。示例性的霉菌毒素包括曲霉产生的黄曲霉毒素。
在一些实施方式中,毒力因子是破坏性酶。破坏性酶是通过破坏宿主细胞和组织而引起毒力的酶。示例性破坏性酶包括但不限于蛋白酶,脂肪酶和dna酶。例如,由细菌(如大肠杆菌和金黄色葡萄球菌)产生的溶血素分解宿主细胞,包括红细胞。
在一些实施方式中,毒力因子是调节宿主gtp酶活性的蛋白质。毒力因子可以充当鸟嘌呤核苷酸交换因子(gef)或gtp酶激活蛋白(gap)来修饰宿主gtp酶的活性,或者共价修饰宿主gtp酶本身。
示例性病毒毒力基因包括控制病毒潜伏期的病毒基因。另外的示例性病毒毒力基因包括有助于逃避宿主免疫应答的病毒基因以及病毒生命周期的各个方面,例如结合和进入宿主细胞或病毒体产生和释放。
在一些实施方式中,毒力因子是染色体的和固有的。例如,细菌毒力因子例如脂多糖(内毒素)是染色体的。
在一些实施方式中,毒力因子由通过水平基因转移传播的流动遗传元件编码。在一些实施方式中,移动遗传元件是质粒,转座元件,噬菌体元件或核酶。在一些实施方式中,在获取毒力因子之前,毒力因子的水平基因转移将作为共生或共生(共生)微生物的微生物转化为病原微生物。
在一些实施方式中,非宿主是病毒,细菌或真菌,并且靶序列包含毒力因子序列。
在一些实施方式中,非宿主是细菌,并且靶序列包含抗生素抗性基因的序列。在一些实施方式中,抗生素抗性基因由通过水平基因转移传播的流动遗传元件编码。在一些实施方式中,移动遗传元件是质粒,转座元件,噬菌体元件或核酶。
在一些实施方式中,抗生素抗性基因传达对喹诺酮类,β-内酰胺类,大环内酯类,四环素类,磺酰胺类,氨基糖苷类或万古霉素类的抗性。
在一些实施方式中,测序样本包括宏基因组学样本。如本文所用,术语“元基因组学”表示例如使用本公开的组合物和方法对混合生物样本进行直接分析。
在一些实施方式中,宏基因组学样本是环境样本。示例性环境样本包括但不限于土壤,水和空气样本。在一些实施方式中,环境样本包括从人造表面,例如从医院的表面获取的样本。
在一些实施方式中,样本包括环境样本,并且靶序列包括毒力因子序列,抗生素抗性基因或与致病性相关的序列。例如,通过侧向基因转移产生抗生素抗性是主要的公共卫生问题。因此,使用本公开的组合物和方法,对从使用抗生素的环境例如医院或农场采集的宏基因组样本中的抗生素抗性基因的频率进行量化是有益的。
在一些实施方式中,本公开的归一化对照被设计成在样本提取和/或文库制备期间模拟测序样本中靶序列即非宿主序列的特性。在一些实施方式中,归一化对照的多核苷酸群是从核酸样本中纯化的,并且该核酸样本是从病毒,细菌,真菌或真核寄生虫中分离或衍生的。在一些实施方式中,从其纯化nc的病毒,细菌,真菌或真核生物与非宿主的病毒,细菌,真菌或真核寄生虫相似但不相同。
在一些实施方式中,非宿主是病毒,并且归一化对照的多核苷酸是从相关病毒或具有与非宿主相似性质的病毒中纯化的。例如,如果非宿主是慢病毒,则从相关物种慢病毒中纯化归一化对照的多核苷酸。在一些实施方式中,病毒是t4噬菌体或巨细胞病毒(cmv)。或者,如果非宿主是细菌,则从相关细菌种类或具有与非宿主性质相似的细菌种类中纯化归一化对照多核苷酸。
nc可以为f一种或多种生物。例如,nc可以包含从一种或多种生物或生物物种(例如病毒,细菌,真菌或真核生物)提取的多核苷酸,其模拟混合的宿主/非宿主样本中非宿主序列的一种或多种特性。
在某些实施方式中,nc用于确定样本中不同核酸的相对量。例如,归一化对照可用于确定来自被感染宿主(例如感染病毒或细菌的人类宿主)样本中病原体(非宿主)的丰度值。可以通过将映射到病原体的ngs读段除以映射到nc的ngs读段的数量来补偿病原体的丰度值,以补偿背景宿主的不同含量。然后可以将此值映射到标准曲线,该曲线以相同的方式生成和标准化,并且可以从标准曲线推断出病原体滴度。使用这样的方法,则不一定需要确定绝对病原体负荷。
在某些实施方式中,nc用于确定样本中不同核酸的绝对量。例如,nc可以用于确定来自被感染宿主的样本(例如,来自被病毒或细菌感染的人类宿主的样本)中病原体(非宿主)的绝对量。可以通过将映射到病原体的ngs读段的数目除以映射到nc来补偿不同背景宿主含量的ngs读段的数目来确定病原体的丰度值。基于该比率和nc的输入量(例如,噬菌体或多核苷酸的拷贝),可以确定相对病原体基因组拷贝。然后可以针对病原体基因组大小校正该值,以在起始材料中得出“绝对”病原体丰度。
在一些实施方式中,从其纯化归一化对照多核苷酸的感兴趣的物种与非宿主物种相似但不相同。
在一些实施方式中,测序样本包括来自已被诊断患有某种癌症的受试者的癌症相关样本。癌症的进展是一个多步骤的过程,需要随着时间的推移积累遗传变化。在一些实施方式中,导致癌症的遗传改变包括激活原癌基因并且使肿瘤抑制基因和dna修复基因失调的遗传改变。导致癌症的遗传变化使以前正常的细胞逃避了细胞分裂的调控,并不受限制地分裂。因此,癌症包括具有不同突变的异质细胞群体。因此,在一些实施方式中,癌症相关样本包括细胞群体,该细胞群体包含相对于受试者的非癌细胞或细胞中的核酸具有相对于核酸至少一种遗传变化的核酸(例如基因,dna或rna)。来自没有癌症的受试者,或来自患有不同类型癌症的受试者。
在一些实施方式中,仅在癌症样本中的异质细胞群体中很少发生感兴趣的突变。但是,癌症样本中的特定突变可预测对治疗的临床反应。因此,使用本公开的组合物和方法鉴定和量化异质癌症样本中突变的发生是有益的。当细胞类型和/或突变很少见时,本本公开的组合物和方法特别有益并且有用。
在一些实施方式中,癌细胞中的至少一种遗传变化包括单核苷酸多态性(snp),插入,缺失,倒位或染色体重排。
在一些实施方式中,至少一种遗传机会与原癌基因的活化或肿瘤抑制基因的失活相关。示例性原癌基因包括但不限于人表皮生长因子受体2(her2),ras,myc原癌基因(myc),src原癌基因(src),端粒酶,bcl-2,细胞凋亡调节剂(bcl-2)和表皮生长因子受体(egfr)。示例性的肿瘤抑制基因包括但不限于肿瘤蛋白p53(p53),成视网膜细胞瘤(rb),腺瘤性息肉病大肠杆菌(apc),乳腺癌敏感性基因1(brca1)和乳腺癌敏感性基因2(brca2)。
在一些实施方式中,靶序列包括在癌症中具有至少一种遗传改变的序列。
在一些实施方式中,从中纯化归一化对照多核苷酸的核酸样本中的核酸序列与测序样本中的靶序列不同。在一些实施方式中,核酸样本中核酸的序列具有小于或等于1%,2%,3%,4%,5%,6%,7%,8%,9%,10的%,11%,12%,13%,14%,15%,16%,17%,18%,19%,20%,30%,40%,50%,60%,70%,80%,与测序样本中的靶序列具有85%,90%,95%,96%,97%,98%或99%的同一性。
浓度
在一些实施方式中,nc被设计为在dna提取期间添加至宿主(例如患者)样本中,以作为非宿主(例如病原体)鉴定管线的一部分对序列读段进行归一化。归一化对照片段共同设计为使得它们代表从样本获得的总序列读段的一小部分。归一化对照片段总体上应该代表从样本获得的序列读段的一小部分(<5%)。在一些实施方式中,归一化对照片段的频率与样本中靶序列的频率匹配。
设想在多核苷酸组之间的浓度的每种可能的排列都在本公开的范围内。
在一些实施方式中,每组的多核苷酸处于相同浓度。例如,在一些实施方式中,在归一化对照组合物中总浓度为3pg/μl,三组中的每组的相同浓度为每微升(μl)1皮克(pg)。
在一些实施方式中,每组的多核苷酸的浓度不同。在一些实施方式中,多核苷酸的每组可以不同的浓度存在。例如,在某些实施方式中,如果三个组以1:2:4的浓度比存在,并且第一组的浓度为1pg/μl,则第二组的浓度为2pg/μl,第三组的浓度为4pg/μl,在归一化对照组合物中的总浓度为7pg/μl。
设想在组内的亚组之间以及在不同组的亚组之间的浓度的每种可能的排列都在本公开的范围内。
在一些实施方式中,至少三个组包括至少一组,其中该组中的至少三个亚组中的多核苷酸的浓度是相等的,以及至少一组,其中该组中的亚组的浓度是不相等的。
在一些实施方式中,至少三个组中的每个中的至少三个亚组中的每个中的多核苷酸的浓度不等于该组中其他亚组的浓度。在一些实施方式中,组中至少三个亚组中每一个的浓度以线性序列或几何序列分布。线性序列是数字序列,其中所述序列中的下一个数字每次相对于前一个数字增加或减少相同的量。通常,线性序列由以下公式表示:
un=dxn+c
其中d是序列中连续项之间的第一个差异,n是序列中的项,并且c是常数。在其中浓度为线性序列的组中存在三个亚组的示例性非限制性实施方式中,这三个亚组的浓度为例如1pg/μl,2pg/μl和3pg/μl。
几何序列是数字序列,其中第一项之后的每个项都可以通过将前一项乘以固定的非零数(称为“公共比率”)来找到。通常,几何序列由下式表示:
un=u1r-1
其中r是公共比率。在一组示例性的非限制性实施方式中,其中组中的三个亚组的浓度是几何序列,则这三个亚组的浓度为例如2pg/μl,4pg/μl和8pg。/μl。每组多核苷酸内亚组的相对浓度是本公开归一化对照的有利特征。在ngs之后,映射回多核苷酸的每个亚组和/或组的测序读段的相对丰度将反映归一化对照组合物以及因此样本中ngs之前的亚组和/或组的相对浓度。因此,计算组或亚组内读段的相对丰度可提供一种内部质量控制措施,并可以为测序样本中靶序列的初始浓度提供更准确的模型。
在一些实施方式中,组中的一个亚组具有基线浓度,并且该组中非基线亚组的浓度是基线的整数倍。在一些实施方式中,组中的一个亚组具有基线浓度,并且该组中每个亚组的浓度是该组中不包括基线的另一亚组的浓度的两倍。在一些实施方式中,该组中的至少三个亚组以1:2:4的浓度比存在。
在一些实施方式中,以允许单个nc序列的浓度的宽动态范围的方式混合多种多核苷酸。浓度范围可以从101到107,某些片段的浓度处于中间水平。使用包括qpcr在内的多种方法对归一化对照进行验证。
在一些实施方式中,在所述归一化对照组合物中,多核苷酸群体的浓度介于约0.005皮克(pg)/微升(μl)与约5000pg/μl之间、介于约0.005pg/μl与约1000pg/μl、之间、介于约0.005pg/μl与约700pg/μl之间、介于约0.005pg/μl与约500pg/μl之间、介于约0.005pg/μl与约100pg/μl之间、介于约0.005pg/μl与约50pg/μl之间、介于约0.05pg/μl与约5000pg/μl之间、介于约0.05pg/μl与约1000pg/μl之间、介于约0.05pg/μl与约700pg/μl之间、介于约0.05pg/μl与约500pg/μl之间、介于约0.05pg/μl与约100pg/μl之间、介于约0.05pg/μl与约50pg/μl之间、介于约0.5pg/μl与约1000pg/μl之间、介于约0.5pg/μl与约700pg/μl之间、介于约0.5pg/μl与约500pg/μl之间、介于约0.5pg/μl与约100pg/μl之间、介于约0.05pg/μl与约50pg/μl之间、介于约5pg/μl与约1000pg/μl之间、介于约5pg/μl与约700pg/μl之间、介于约5pg/μl与约500pg/μl之间、介于约5pg/μl与约100pg/μl之间或介于约5pg/μl与约50pg/μl之间。在一些实施方式中,在归一化对照组合物中,多核苷酸群体的浓度介于约0.005皮克(pg)/微升(μl)与500pg/μl之间。在一些实施方式中,在归一化对照组合物中多核苷酸群体的浓度介于约0.5pg/μl与约50pg/μl之间。在一些实施方式中,在归一化对照组合物中多核苷酸群体的浓度介于约10pg/μl与约20pg/μl之间。在一些实施方式中,在归一化对照组合物中,多核苷酸群体的浓度介于约1pg/μl与约2pg/μl之间。在一些实施方式中,在归一化对照组合物中,多核苷酸群体的浓度为介于约0.1pg/μl与约0.2pg/μl之间。在一些实施方式中,在归一化对照组合物中,多核苷酸群体的浓度为1.4pg/μl,包括端点在内。
索引编制
在一些实施方式中,归一化对照多核苷酸包含编码唯一分子标识符的序列。在一些实施方式中,编码唯一分子标识符的序列包括第一索引序列或第二索引序列。
本公开的示例性非限制性索引策略使用两个索引,第一索引和第二索引。在一些实施方式中,编码独特分子标识符的序列包括共同的引物序列,索引序列和衔接子序列。在一些实施方式中,共同引物序列包含ngs的流动池附着位点。
在一些实施方式中,索引序列是独特的4-20bp序列。在一些实施方式中,索引序列是独特的6bp、8bp或10bp序列。在一些实施方式中,索引序列是独特的6bp序列。索引的长度将取决于文库的复杂性。合适的索引序列长度对于本领域技术人员将是显而易见的。在一些实施方式中,包含第一引物序列、第一索引序列和第一衔接子序列的序列附接至多核苷酸的5'端,并且包含第二引物序列、第二索引序列和第二衔接子的序列序列附接到同一多核苷酸的3'端。所述多核苷酸是例如本公开的归一化对照组合物的多核苷酸。在一些实施方式中,每组多核苷酸在该组中的多个多核苷酸的5'和3'末端上包含唯一的双索引。在该实施方式中,双索引对于该组内的所有多个多核苷酸是相同的,并且对于组之间的多核苷酸是不同的。在一些实施方式中,多核苷酸的每个亚组在该亚组中的多个多核苷酸的5'和3'末端上包含唯一的双索引。在该实施方式中,双索引对于亚组内的所有多个多核苷酸是相同的,但是对于不同亚组中的多核苷酸是不同的。在一些实施方式中,每个组内和每个亚组内的多个多核苷酸具有对该组和/或亚组特异的独特的双重索引。
本公开的非限制性示例性双索引系统是illuminai5和i7双索引系统。
在一些实施方式中,归一化对照被设计为测量dna或rna的转化。将测序样本中的靶序列靶向可测序的文库分子。在一些实施方式中,在文库制备期间起始材料到终止材料的转化率很差,并且难以在所有样本中可靠地测量。通过双索引nc多核苷酸的末端,可以跟踪在文库制备过程中的任何时间转换的分子以及每个样本的转换方式。这允许计算样本类型不可知库转换因子。这也允许将质量控制内置到样本制备中。使用此技术,可以比较试剂批次,酶促反应的效率以及影响ngs的许多其他指标。这些比较可以按照测序反应的步骤进行,通常需要等到文库制备过程结束才能确定是否出现问题。
蛋白质
归一化对照多核苷酸包含脱氧核糖核酸(dna)分子,核糖核酸(rna)分子或dna-rna杂合分子。
与蛋白质的结合是从ngs读段计数计算靶序列浓度时变异和错误的重要来源。例如,在一些实施方式中,靶序列是病毒序列,并且包含病毒序列的多核苷酸与衣壳的缔合防止了在样本和文库制备期间靶序列的纯化和/或片段化。因此,将蛋白质添加到本本公开的归一化对照组合物中以模拟测序样本中靶序列的核酸/蛋白质相互作用,从而增加了nc在ngs期间模拟靶序列行为的能力。
在本公开的归一化对照的一些实施方式中,归一化对照还包含至少一种蛋白质。在一些实施方式中,至少一种蛋白质是分离或衍生自细菌,病毒或真核寄生虫。在一些实施方式中,至少一种蛋白是病毒衣壳蛋白或细菌细胞壁蛋白。示例性但非限制性的病毒衣壳蛋白包括分离或衍生自腺病毒,腺伴随病毒(aav),慢病毒或逆转录病毒的病毒衣壳蛋白。
在一些实施方式中,多核苷酸群和蛋白质可操作地连接。在示例性的实施方式中,蛋白质是病毒衣壳蛋白,并且多核苷酸被包封在病毒衣壳蛋白内。在其他实施方式中,蛋白质和多核苷酸被结合。
人工序列
在一些实施方式中,归一化对照多核苷酸包含启动子序列。在一些实施方式中,启动子序列包含选自t7启动子序列,sp6启动子序列或t3启动子序列的启动子序列。在一些实施方式中,特别是其中启动子是t7启动子的那些实施方式,t7启动子包含序列5'-taatacgactcactatagg-3'(seqidno:14)。在一些实施方式中,t7启动子包含序列5'-taatacgactcactataggg-3'(seqidno:15)。在一些实施方式中,t7启动子包含序列5'-gcctcgagctaatacgactcactatagag-3'(seqidno:16)。在一些实施方式中,sp6启动子包含序列5'-atttaggtgacactatag-3'((seqidno:17)。在一些实施方式中,sp6启动子包含序列5'-catacgatttaggtgacactatag-3(seqidno:18)。在一些实施方式中,t3启动子包含序列5'aattaaccctcactaaag3'的(seqidno:19)。
在一些实施方式中,归一化对照多核苷酸包含另外的引物序列。引物序列可以位于组分和/或分离的序列的5'和3'。引物序列可以作为组分序列的计算机设计的一部分被添加,或者其次被添加到组分和/或分离的序列中。引物序列可以通过例如衔接子连接或组分和/或分离的序列的随机引物和延伸而添加到组分和/或分离的序列。
混合组合物
在一些实施方式中,本公开内容的归一化对照包括合成多核苷酸和已分离或衍生自一种或多种生物的多核苷酸的混合物。例如,样本中的一组靶序列可以覆盖一定范围的长度,该范围超出了合成多核苷酸的当前制造方法的长度(例如,10kb的长度)。在该示例中,为了用归一化对照完全模拟靶序列的长度范围,归一化对照包括组分和分离的多核苷酸的混合物。
在一些实施方式中,使用本公开的方法从生物中纯化归一化对照中的至少一组多核苷酸。在一些实施方式中,生物体是病毒、细菌、真菌或真核寄生虫。在一些实施方式中,生物体与混合宿主/非宿主测序样本中的非宿主共享一个或多个特征。在一些实施方式中,病毒是噬菌体t4。
在一些实施方式中,还以已知浓度将噬菌体t4添加或添加到基于合成的多核苷酸的nc(基于寡核苷酸的nc)中。
将噬菌体t4添加到归一化对照中可以使归一化对照来控制提取变异。噬菌体t4的额外好处是,与基于寡核苷酸的nc相比,它更便宜且更易于制造,尤其是对于长序列而言。
制备归一化对照的方法
从头合成
本公开内容提供了进行归一化对照的方法,所述归一化对照包括至少三组多核苷酸,其中每组内的多核苷酸具有相同的长度。在一些实施方式中,至少三组多核苷酸中的每一个进一步包含多核苷酸的至少三个亚组。
在一些实施方式中,至少三个组包含相同序列的多核苷酸。在一些实施方式中,至少三个组包含序列不同的多核苷酸
在本公开的方法的一些实施方式中,所述方法包括合成每个独特组和/或亚组的多核苷酸作为寡核苷酸,对每个独特组和/或亚组的浓度进行定量,以及混合预定量的每个独特组和/或亚组以产生nc组合物。
在一些实施方式中,多核苷酸是dna多核苷酸。在一些实施方式中,多核苷酸在约10bp至约3000bp之间。在一些实施方式中,多核苷酸在约10bp至约1200bp之间。在一些实施方式中,多核苷酸在10bp与250bp之间。在一些实施方式中,多核苷酸在约150bp和约1500bp之间。在一些实施方式中,多核苷酸在约400bp和约1100bp之间。在一些实施方式中,多核苷酸在约500bp至约1500bp之间。
在一些实施方式中,多核苷酸具有与测序样本中的靶序列相同的至少一种性质。预期靶序列的所有序列性质都在本公开的归一化对照的范围内。在一些实施方式中,靶序列包含转座元件序列,病毒序列,细菌序列,真菌序列,真核寄生虫序列或一种或多种人类基因的序列。在一些实施方式中,至少一种特性包括转座元件序列、病毒序列、细菌序列、真菌序列、真核寄生虫序列、或一个或多个人类基因序列的序列特性。
在一些实施方式中,归一化对照的多核苷酸包含rna。在一些实施方式中,所述dna多核苷酸进一步包含编码启动子的序列,并且所述方法进一步包括体外转录每个dna多核苷酸以产生多个rna,定量所述多个rna中的每个的浓度,以及在所述rna中混合多个rna。预定量以产生归一化对照。示例性的启动子包括但不限于t3,sp6和t7。
设想在多核苷酸的组和亚组之间的浓度的每种可能的排列都在本公开的范围内。
设想在本组的范围内,在一个组内的各亚组之间以及在不同组的各亚组之间的浓度的每种可能的布置。
浓度可以表示为每体积的重量(例如,象形图(每微升pg(pg/微升)或每μl纳克(ng)))或每体积的多核苷酸分子数(摩尔浓度)。对于本领域的普通技术人员而言,如何转换浓度的度量将是显而易见的。
在一些实施方式中,将每个多核苷酸合成为dna寡核苷酸,并且任选地,在体外转录以产生多个rna多核苷酸。定量每个组或亚组的多个多核苷酸中的每一个多核苷酸,并以本公开的浓度比混合多个多核苷酸。然后将最终的归一化对照组合物稀释至介于约0.001皮克(pg)/微升(μl)与约5000pg/μl之间、介于约0.005pg/μl与约1000pg/μl之间、介于约0.005pg/μl与约700pg/μl之间、介于约0.005pg/μl与约500pg/μl之间、介于约0.005pg/μl与约100pg/μl之间、介于约0.005pg/μl与约50pg/μl之间、介于约0.05pg/μl与5000pg/μl之间、介于约0.05pg/μl与约1000pg/μl之间、介于约0.05pg/μl与约700pg/μl之间、介于约0.05pg/μl与约500pg/μl之间、介于约0.05pg/μl与约00pg/μl之间、介于约0.05pg/μl与约50pg/μl之间、介于约0.5pg/μl与约1000pg/μl之间、介于约0.5pg/μl与约700pg/μl之间、介于约0.5pg/μl与约500pg/μl之间、介于约0.5pg/μl与约100pg/μl之间、介于约0.05pg/μl与约50pg/μl之间、介于约5pg/μl与约1000pg/μl之间、介于约5pg/μl与约700pg/μl之间、介于约5pg/μl与约500pg/μl之间、介于约5pg/μl与约100pg/μl,或介于约5pg/μl与约50pg/μl之间的浓度。在一些实施方式中,归一化对照的浓度介于约0.005pg/μl与约500pg/μl之间。在一些实施方式中,归一化对照的浓度介于约0.5pg/μl与约50pg/μl之间。在一些实施方式中,归一化对照的浓度介于约10pg/μl与约20pg/μl之间。在一些实施方式中,归一化对照的浓度介于约1pg/μl与约2pg/μl之间。在一些实施方式中,归一化对照的浓度介于约0.1pg/μl与约0.2pg/μl之间。在一些实施方式中,归一化对照的浓度为1.4pg/μl。
本公开提供了制备多个归一化对照寡核苷酸的方法,所述方法包括:(a)从至少一个参考序列产生多个参考序列片段;(b)根据参考序列片段的数量产生至少一个参数的分布;(c)将所述分布划分成箱;(d)从所述箱中的至少子集中选择至少一个参考序列片段;(e)改组参考序列以产生经改组的序列;以及(f)合成包含经改组的序列的寡核苷酸;从而产生多个归一化对照寡核苷酸。
本公开提供了制备多个归一化对照寡核苷酸的方法,所述方法包括:(a)使用滑动窗从至少一个参考序列产生多个参考序列片段;(b)根据参考序列片段的数量产生至少一个参数的分布;(c)将所述分布划分成箱;(d)从所述箱中的至少子集中选择至少两个参考序列片段,其中所述至少两个参考序列片段在参考序列中是不连续的,或者是从不同的参考序列中选择的;(e)将来自至少3个箱中的每一个箱的至少两个参考序列片段连接起来;以及(f)合成包含连接的参考序列片段的寡核苷酸;从而产生多个归一化对照寡核苷酸。在一些实施方式中,滑动窗包括1个碱基对(bp)、2bp、3bp、4bp或5bp的滑动窗。在一些实施方式中,所述参考序列片段是对应参考序列的约15-60个、约20-40个、约20-30个、约15-32个、20-32个或约25-35个连续核苷酸。在一些实施方式中,参考序列片段是对应参考序列的29、30、31、32、33或34个碱基对。在一些实施方式中,参考序列片段小于测序读段的平均长度的一半。
在一些实施方式中,该参数包括以下各项中的至少一项:(1)gc含量百分比、(2)熵、(3)复杂性、(4)eiip,或它们的组合。在一些实施方式中,该参数包括gc含量百分比。
根据归一化对照的所需分辨率,可以将分布(例如gc含量的分布)划分为任意数量的合适的箱。例如,每个箱可以代表所述分布的1%、2%、5%、10%、15%、或20%。参考序列片段可以从每个箱或从所述箱的子集中选择。参考序列片段选自来自所述gc含量百分比分布的第10、第30、第50、第70、第80和第90百分位箱。
在一些实施方式中,步骤(d)包括从每个箱中选择至少2个参考序列片段。例如,从每个箱中选择至少2、3、4、5、6、7、8、9或10个参考序列片段。在一些实施方式中,步骤(e)还包括连接来自每个箱的经改组的参考序列。在一些实施方式中,仅连接来自给定箱的序列,以模拟对应箱中的参考序列片段的特性,并因此对沿分布的多个点建模。
在一些实施方式中,所述至少一个参考序列包含至少2个、至少10个、至少20个、至少50个、至少100个、至少200个、至少300个、至少400个、至少500个、至少600个、至少700个、至少800个、至少900个、至少1,000个、至少1,200个、至少1,300个、至少1,400个、至少1,500个、至少1,600个、至少1,700个、至少1,800个、至少1,900个、至少2,000个、至少2,200个、至少2,400个、至少2,600个、至少2,800个、至少3,000个、至少4,000个、至少5,000个、至少6,000个、至少7,000个、至少8,000个、至少9,000或至少10,000个参考序列。在一些实施方式中,至少一个参考序列包含约2,000个参考序列。在一些实施方式中,至少一个参考序列包含基因组序列或cdna序列或它们的组合。例如,对应于第一生物的参考序列可以是基因组序列,并且对应于第二生物的参考序列可以是cdna序列。或者,多种生物(所有生物)的参考序列可以是基因组序列或cdna序列。
在一些实施方式中,参考序列片段是参考序列的介于约10-100个之间、介于约10-80个之间、介于约10-70个之间、介于约10-60个之间、介于约10-50个之间、介于约10-40个之间、介于约10-30个之间、介于约10-20个之间、介于约20-100个之间、介于约20-80个之间、介于约20-60个之间、介于约20-50个之间、介于约20-40个之间、介于约20-30个之间、介于约10-35个之间、介于约20-35个之间、或介于约20-25个之间的连续核苷酸。在一些实施方式中,所述参考序列片段是参考序列的介于约15-60个之间、介于约20-40个之间、介于约20-30个之间、介于约15-32个之间、介于20-32个个之间或介于约25-35个之间的连续核苷酸。在一些实施方式中,参考序列片段包含参考序列的20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、31个、32个、33个、34个、35个、36个、37个、38个、39个、40个、41个、42个、43个、44个、45个、46个、47个、48个、49个、50个、51个、52个、53个、54个、55个、56个、57个、58个、59个、60个、61个、62个、63个、64个或65个连续碱基对。
在一些实施方式中,每个归一化对照寡核苷酸具有以下中的至少三者:与来自参考序列分布中的对应箱的参考序列片段(1)类似的gc含量百分比、(2)类似的熵、(3)类似的eiip、(4)类似的复杂性、或(5)类似的长度。
多重合成
在一些实施方式,归一化对照包括合成dna多核苷酸的混合物。
本公开提供了制备合成多核苷酸群体的方法,所述方法包括:(a)合成至少三个dna分子群体,其中所述至少三个群体中的每个群体内的每个dna分子具有相同的序列,并且其中所述dna分子中的每个dna分子的序列从5'至3'包含第一组分序列、第一限制性酶切位点、第二组分序列、第二限制性位点和第三组分序列;(b)对溶液中dna分子群体中的每个群体的量进行定量;(c)用切割第一和第二限制性位点的限制性酶消化所述至少三个dna分子群体中的每个群体,以产生至少三个经消化的dna分子群体;以及(d)将预定量的所述至少三个经消化的dna分子群体中的各个群体在单个组合物中混合。在一些实施方式中,所述至少三个合成dna分子中的每一个合成dna分子包含与所述至少三个合成dna分子中的任何其他合成dna分子的第一、第二和第三组分序列具有相同长度的第一、第二和第三组分序列。
在本公开的方法的一些实施方式中,第一、第二和第三组分序列均介于约15bp与约3000bp之间。在一些实施方式中,第一、第二和第三组分序列均介于约15bp与约1200bp之间。在一些实施方式中,第一、第二和第三组分序列均介于约100bp与约600bp之间。在一些实施方式中,第一、第二和第三组分序列均介于约150bp与约500bp之间。在一些实施方式中,第一、第二和第三组分序列分别是175bp、250bp和450bp。
在一些实施方式中,至少三个dna分子中的任何一个dna分子的第一、第二和第三组分序列都不是相同的序列。在一些实施方式中,第一、第二和第三组分序列是随机序列。在一些实施方式中,使用随机序列生成器来生成随机序列。可以在www.bioinformatics.org/sms2/random_dna.html找到示例性但非限制性的随机序列生成器,并且通过引用将其并入本文。在一些实施方式中,随机序列不映射到ncbi核苷酸数据库中的任何生物体的基因组。在一些实施方式中,可以设计包括序列的随机序列。重要的序列属性(例如gc含量),并排除其他(例如,常见的限制性酶切位点)。
在一些实施方式中,至少三个dna分子中的每一个的第一,第二和第三组分序列具有与测序样本中的靶序列相同的至少一个特性。
在一些实施方式中,第一和第二限制性位点的序列是相同的。在一些实施方式中,限制性位点是被ii型限制性核酸内切酶识别和切割的限制性位点。合适的限制性位点的选择将取决于第一、第二和第三组分序列,并且对于本领域普通技术人员而言将是显而易见的。在一些示例性实施方式中,选择在第一、第二和第三组分序列中的任何一者中均不出现的限制性位点。
在一些实施方式中,所述第一和第二限制位点的序列选自由以下项组成的组:ecori位点、bamhi位点、hindiii位点、noti位点、pvuii位点、smai位点、ecorv位点、kpni位点、psti位点、sali位点、scai位点、spei位点、sphi位点、stui位点、和xbai位点的序列。在一些实施方式中,用于消化至少三个合成dna分子的至少三个群体中的每一个群体的限制性酶包括ecori、bamhi、hindiii、noti、pvuii、smai、ecorv、kpni、psti、sali、scai、spei、sphi、stui或xbai。在一些实施方式中,第一和第二限制性位点的序列包含xhoi位点,而限制性酶包含xhoi。
在一些实施方式中,至少三个dna分子中的每一个dna分子的序列还包含第一引物序列和与第二引物序列互补的序列,所述第一引物序列位于第一随机序列的5',并且与所述第二引物序列互补的序列位于第三随机序列的3'。在一些实施方式中,第一引物序列包含序列5'-tgaagaactgcggcagg-3'(seqidno:12)(mito阵列引物)。在一些实施方式中,第二引物序列包含序列5'-agctggaagtgcagacc-3'(ribo阵列引物序列),与第二引物序列互补的序列包含序列5'-ggtctgcacttccagct-3'的(seqidno:13)。
在一些实施方式中,本公开的方法包括对溶液中至少三个dna分子中的每一个dna分子的量进行定量。在一些实施方式中,本公开的方法进一步对溶液中至少三个经消化的dna分子群体中的每一个群体的量进行定量。对溶液中每个dna分子群体的量进行定量和/或对溶液中至少三个经消化的dna分子群体中的每一个群体的量进行定量的方法对于本领域普通技术人员来说将是显而易见的。dna定量方法的非限制性示例包括实时pcr、液滴数字pcr(ddpcr)和分光光度法。
在一些实施方式中,本公开的方法包括将预定量的至少三个经消化的dna分子群体中的每一个群体在单个组合物中混合。
在一些实施方式中,至少三个经消化的dna分子群体的预定量是相等的。在一些实施方式中,其中至少三个经消化的dna分子群体的预定量不相等。在一些实施方式中,至少三个经消化的dna分子群体的预定量是线性序列或几何序列。
在一些实施方式中,一个数量的消化的合成dna分子的预定量是基线,并且至少两个另外的数量的消化的dna分子的预定量是基线的整数倍。在一些实施方式中,一个消化的dna分子群体的预定量是基线,并且其中每个另外的消化的dna分子群体的预定量是另一个消化的dna分子群体的预定量,不包括基线。
在一些实施方式中,至少三个消化的合成dna分子群体的预定量为1:2:4的比例。
在示例性实施方式中,将三个群体的消化的dna分子以a:b:c的比例混合,其中“a”,“b”和“c”代表任何预定的浓度比例(例如1:2:4、1:3:5或任何线性或几何顺序)。在该实施方式中,三个dna分子中的每个的第一组分序列是相同的长度,三个dna分子中的每个的第二组分序列是相同的长度,并且三个合成dna分子中的每个的第三组分序列是相同的长度。相同的长度。所得的nc组成由三组组成,每组由三个亚组组成。第一组的每个亚组由包含每个dna分子的第一组分序列的多核苷酸组成,第二组的每个亚组由三个dna分子中的每个包含第二组分序列的多核苷酸组成,第三组的每个亚组包括包含第三组分的多核苷酸序列每个dna分子。当三个消化的dna分子群体以a:b:c的比例混合时,第一组三个亚组的三种多核苷酸以a:b:c的比例存在。由于每个消化的dna分子群体对每个组的贡献均等,因此每个组以相同的浓度存在于组合物中。
体外转录
在一些实施方式中,归一化对照包括rna多核苷酸。本公开提供了进行这种rna归一化对照的方法。
本公开内容提供了体外转录方法,以转录为聚合酶的启动子序列的3'的序列(例如,t3,sp6或t7聚合酶)。设想具有5'启动子序列的任何序列是用于本公开内容的体外转录方法的合适模板。
本公开内容提供了进行归一化对照的方法,包括合成至少三个dna多核苷酸,每个多核苷酸包含启动子序列和组分序列,在体外转录每个多核苷酸以产生多个rna,定量多个中的每个的浓度混合rna,并以预定量混合多个rna以产生nc组合物。
本公开内容提供了制备合成核酸分子群体的方法,其包括:(a)合成至少三组dna分子;其中每个dna分子包含5'至3'的第一组分序列,启动子序列,第一限制性酶切位点,第二组分序列,第二启动子序列,第二限制性位点,第三组分序列第三启动子序列;(b)用限制性酶消化每一组dna分子,所述限制性酶切割第一和第二限制性酶切位点,以产生至少三个被消化的合成dna分子群体;(c)在体外转录每组dna分子以产生至少三个rna分子种群;(d)定量每个rna分子群体中产生的rna数量;(e)混合预定量的每个rna分子群体以产生单一的归一化组合物。
在一些实施方式中,每个dna分子包含与任何其他dna分子的第一,第二和第三组分序列相同长度的第一,第二和第三组分序列。
在一些实施方式中,第一,第二和第三组分序列各自在约15bp和约1200bp之间。在一些实施方式中,第一、第二和第三组分序列均介于约150bp与约500bp之间。在一些实施方式中,第一、第二和第三组分序列均介于约100bp与约600bp之间。
在一些实施方式中,任何dna分子的第一,第二和第三组分序列都不是相同序列。在一些实施方式中,每个dna分子的第一,第二和第三组分序列是随机序列。在一些实施方式中,每个dna分子的第一,第二和第三组分序列具有与样本中的靶序列相同的至少一个特性。
本公开提供了用限制性酶消化至少三个合成dna分子中的每一个的方法,所述限制性酶例如ii型限制核酸内切酶,其切割第一和第二限制性酶切位点以产生至少三个被消化的dna分子群体。
在一些实施方式中,第一和第二限制位点的序列是相同的。在一些实施方式中,限制性位点是被ii型限制性核酸内切酶识别和切割的限制性位点。合适的限制性位点的选择将取决于第一、第二和第三组分序列,并且对于本领域普通技术人员而言将是显而易见的。在一些示例性实施方式中,选择在第一、第二和第三组分序列中的任何一者中均不出现的限制性位点。
在一些实施方式中,第一和第二限制性位点的序列选自ecori位点,bamhi位点,hindiii位点,noti位点,pvuii位点,smai位点,ecorv位点的序列。kpni站点,psti站点,sali站点,scai站点,spei站点,sphi站点,stui站点和xbai站点。在一些实施方式中,用于消化至少三个合成dna分子的至少三个群体中的每一个群体的限制性酶包括ecori、bamhi、hindiii、noti、pvuii、smai、ecorv、kpni、psti、sali、scai、spei、sphi、stui或xbai。在一些实施方式中,第一和第二限制性位点的序列包含xhoi位点,而限制性酶包含xhoi。
在一些实施方式中,所得的消化的dna分子群体包含具有用于体外转录的5'启动子和组分序列的dna分子。例如,至少三个合成dna分子的每个群体在消化时产生三个dna片段,每个片段包含可操作地连接至第一,第二或第三组分序列的5'启动子。这些dna片段然后可以用作体外转录的模板以产生本公开的rna多核苷酸。在一些实施方式中,在体外转录之前分别纯化和分离片段。在一些实施方式中,片段库用作体外转录的模板。
本公开提供了体外转录每个dna分子群体以产生至少三个rna分子群体的方法。在一些实施方式中,第一启动子序列,第二启动子序列和第三启动子序列包含相同的启动子序列。在一些实施方式中,启动子序列包含选自t7启动子序列,sp6启动子序列或t3启动子序列的启动子序列。在一些实施方式中,特别是在其中启动子是t7启动子的那些实施方式中,t7启动子包含序列5'-taatacgactcactatagg-3'(seqidno:14)。在一些实施方式中,t7启动子包含序列5′-taatacgactcactataggg-3′(seqidno:15)。在一些实施方式中,t7启动子包含序列5'-gcctcgagctaatacgactcactatagag-3'(seqidno:16)。在一些实施方式中,sp6启动子包含序列5'-atttaggtgacactatag-3'(seqidno:17)。在一些实施方式中,sp6启动子包含序列5'-catacgatttaggtgacactatag-3(seqidno:18)。在一些实施方式中,t3启动子包含序列5'aattaaccctcactaaag3'(seqidno:19)。
本公开的聚合酶可以是rna聚合酶ii或rna聚合酶iii聚合酶。在一些实施方式中,聚合酶是t7聚合酶、sp6聚合酶或t3聚合酶。本公开的rna聚合酶可以是野生型聚合酶、组分聚合酶,或已被优化或工程化(例如,用于体外转录)的聚合酶。对于给定的启动子序列(例如,用于t7启动子的t7聚合酶、用于sp6启动子的sp6聚合酶或用于t3启动子的t3聚合酶),本本公开的聚合酶的活性可以是高度特异性的。
t7启动子被t7噬菌体rna聚合酶识别并支持其转录。本公开的t7聚合酶可以是已被优化或工程改造(例如,用于体外转录)的野生型t7聚合酶、组分t7聚合酶或t7聚合酶。t7聚合酶是dna依赖性rna聚合酶,其可催化dna模板在5'至3方向上形成rna。dna模板可以是双链或单链的。t7聚合酶对t7启动子表现出高特异性,可以在体外产生强大的转录,并且能够将修饰的核苷酸(例如,标记的核苷酸)掺入新生的rna转录物中。t7聚合酶的这些特征使其成为用于合成本公开内容的rna的优异的聚合酶。
本公开提供了定量在至少三个合成rna分子群体的每一个中产生的rna的量的方法。定量rna的方法对于本领域普通技术人员将是显而易见的。定量rna的非限制性示例性方法包括分光光度法或用染料例如溴化乙锭进行荧光标记。
本公开提供了混合预定量的每个rna分子群体以产生单一组合物的方法。
在一些实施方式中,每个rna分子群体的预定量是相等的。在一些实施方式中,每个rna分子群体的预定量不相等。在一些实施方式中,预定量的rna分子群体为线性序列或几何序列。
在一些实施方式中,一个rna分子群体的预定量是基线,并且其中另外的rna分子群体的预定量是基线的整数倍。在一些实施方式中,一个rna分子群体的预定量是基线,并且其中每个另外的rna分子群体的预定量是不包括基线的另一rna分子群体的浓度的两倍。在一些实施方式中,至少三个rna分子群体的预定量为1:2:4的比例。
在一些实施方式中,该方法进一步包括将归一化对照稀释至介于约0.001pg/μl与约500pg/μl之间的浓度。在一些实施方式中,该方法进一步包括将归一化对照稀释至介于约0.5pg/μl与约500pg/μl之间的浓度。在一些实施方式中,该方法进一步包括将归一化对照稀释至介于约0.5pg/μl与约50pg/μl之间的浓度。在一些实施方式中,所述方法进一步包括将归一化对照稀释至介于约10pg/μl与约20pg/μl之间的浓度。在一些实施方式中,该方法进一步包括将归一化对照稀释至介于约1.0pg/μl与约2.0pg/μl之间的浓度。在一些实施方式中,该方法进一步包括将归一化对照稀释至介于约0.1pg/μl与约0.2pg/μl之间的浓度。在一些实施方式中,归一化对照的浓度为1.4pg/μl。
分离的序列
本公开提供了制备归一化对照组合物的方法,其中所述归一化对照包含分离的序列。在一些实施方式中,分离的序列是从天然存在的序列中分离的,而没有另外的步骤,例如序列改组或与另外的分离的或组分序列的连接。
因此,本本公开提供了进行归一化对照的方法,所述方法包括:(a)从样本中提取dna;(b)用限制性酶消化dna以产生dna片段集合;(c)分离dna片段集合;(d)从dna片段集合中纯化至少三个dna片段,以产生至少三组多核苷酸,其中所述至少三组多核苷酸中的每个组均包含多个多核苷酸;以及(e)将预定量的至少三组多核苷酸中的每个组混合以产生组合物。
本公开提供了从样本提取dna的方法。在一些实施方式中,样本分离自或衍生自病毒,细菌,真菌或真核寄生虫。在一些实施方式中,样本包含克隆载体。在一些实施方式中,克隆载体是细菌人工染色体(bac),酵母人工染色体(yac),粘粒,粘粒或质粒。在一些实施方式中,样本是质粒。在一些实施方式中,将归一化对照序列从感兴趣的物种例如病毒,细菌或真核寄生虫克隆到克隆载体中,并从产生克隆载体的细胞培养物中提取dna。在一些实施方式中,细胞是细菌(例如大肠杆菌(escherichiacoli))。dna提取的示例性方法包括试剂盒(例如qiagenminiprep)或基于酚的提取方法。合适的dna提取方法对于本领域普通技术人员将是显而易见的。
本公开提供了用限制性酶消化dna以产生dna片段集合的方法。在一些实施方式中,限制性酶选自由以下项组成的组:ecori、bamhi、hindiii、noti、pvuii、smai、ecorv、kpni、psti、sali、scai、spei、sphi、stui和xbai。在一些实施方式中,限制性酶选自由以下项组成的组:ecori、bamhi、hindiii、pvuii、smai、ecorv、kpni、psti、sali、scai、spei、sphi、stui、xbai、noti、asci、fsei、paci、pmei、bglii、bstbi、hincii和sgfi。
在一些实施方式中,通过用限制性酶消化dna而产生的片段介于约15bp与约100kb之间。在一些实施方式中,片段介于约15bp与约50kb之间。在一些实施方式中,片段介于约50bp与约50kb之间。在一些实施方式中,片段介于约500bp与约50kb之间。在一些实施方式中,片段介于约1000bp与约50kb之间。在一些实施方式中,至少三个不同组的多核苷酸中的每一个组的最长是至少15kb的长度、至少20kb的长度、至少25kb的长度、30kb的长度、至少35kb的长度、或至少40kb的长度。
本公开提供了从dna片段集合中纯化至少三个dna片段以产生至少三组多核苷酸的方法,其中至少三组多核苷酸中的每一个都包含多个多核苷酸。在一些实施方式中,分离dna片段集合包括基于片段大小,片段电荷或其组合的分离。在一些实施方式中,分离dna片段集合包括基于片段大小的分离。在一些实施方式中,基于大小分离dna片段包括凝胶电泳,色谱法或切向流过滤(tff)。
在一些实施方式中,基于大小分离dna片段包括凝胶电泳。凝胶电泳根据其大小分离多核苷酸。将多核苷酸样本装入凝胶一端的孔中,并施加电流以使其通过凝胶。多核苷酸带负电,因此向正极移动。多核苷酸的大小决定了其通过凝胶孔迁移的速率。可以通过改变凝胶的琼脂糖浓度(例如,介于在0.8%与3%之间的琼脂糖)来分离大小不同的多核苷酸。或者,可以使用丙烯酰胺凝胶分离较小的多核苷酸。在一些实施方式中,特别是其中分离了大的多核苷酸的那些实施方式,改变电磁场的角度,强度和持续时间可以帮助分离。在一些实施方式中,凝胶电泳是脉冲场凝胶电泳。
在一些实施方式中,基于大小分离dna片段包括色谱法。一种示例性的色谱方法包括阴离子交换色谱。阴离子交换色谱法使用含有带正电基团的离子交换树脂根据多核苷酸的电荷分离多核苷酸,该基团与带负电的分子结合。多核苷酸与柱或基质结合的紧密度基于多核苷酸的负电荷的整体强度,因此取决于大小。
在一些实施方式中,基于大小分离dna片段包括tff。tff是基于膜的过滤过程。基于膜的孔隙率,可以将其分类为微滤或超滤过程。超滤膜的孔径通常在0.001至0.1μm之间。切向流过滤(tff),也称为错流过滤,使进料流平行于膜表面。一部分物流通过膜(渗透物),而其余部分(截留物)再循环回到进料贮存器。多核苷酸通过穿过膜的孔而分离。
在一些实施方式中,所述方法进一步包括将衔接子与至少三个不同组的多核苷酸的每个多核苷酸的至少一个末端连接。
在一些实施方式中,所述方法进一步包括将衔接子连接至所述至少三个不同组的多核苷酸的每个末端。
在一些实施方式中,归一化对照中的至少三个不同组的多核苷酸的序列进一步包含合成的多核苷酸序列。示例性但非限制性的合成序列包括衔接子序列,用于pcr扩增的引物,用于体外转录的启动子序列以及在ngs过程中用于流通池附着的结合位点。
在一些实施方式中,衔接子序列包含编码独特分子标识符的序列。在一些实施方式中,编码唯一分子标识符的序列包括第一索引序列或第二索引序列。本公开的示例性非限制性索引策略使用两个索引,第一索引和第二索引。在一些实施方式中,编码独特分子标识符的序列包括共同的引物序列,索引序列和衔接子序列。在一些实施方式中,共同引物序列包含ngs的流动池附着位点。
在一些实施方式中,索引序列是独特的4-20bp序列。在一些实施方式中,索引序列是独特的6bp、8bp或10bp序列。在一些实施方式中,索引序列是独特的6bp序列。索引的长度将取决于文库的复杂性。合适的索引序列长度对于本领域技术人员将是显而易见的。在一些实施方式中,包含第一引物序列、第一索引序列和第一衔接子序列的序列附接至多核苷酸的5'端,并且包含第二引物序列、第二索引序列和第二衔接子的序列序列附接到同一多核苷酸的3'端。所述多核苷酸是例如本公开的归一化对照组合物的多核苷酸。在一些实施方式中,每组多核苷酸在该组中的多个多核苷酸的5'和3'末端上包含唯一的双索引。在该实施方式中,双索引对于该组内的所有多个多核苷酸是相同的,并且对于组之间的多核苷酸是不同的。
在一些实施方式中,合成序列包含启动子序列。在一些实施方式中,启动子序列包含选自t7启动子序列,sp6启动子序列或t3启动子序列的启动子序列。
分离与从头合成相结合
本公开提供了使用本公开的方法制备归一化对照组合物的方法,其中归一化对照既包含包含合成的序列的多核苷酸,也包含包含从样本中分离的序列的多核苷酸。
本公开内容提供了进行归一化对照的方法,包括:(a)合成至少三组dna分子,其中一组内的dna分子具有相同的序列,并且其中每个dna分子的序列包括5个以上的'至3',第一成分序列,第一限制性酶切位点,第二成分序列,第二限制位点和第三成分序列;(b)定量溶液中每组dna分子的数量;(c)用限制性酶消化每一组dna分子,所述限制性酶切割第一和第二限制位点以产生至少三组消化的dna分子;(d)将预定量的每组经消化的dna分子混合,从而产生单一的归一化对照组合物。
在一些实施方式中,(d)的混合步骤还包括将预定量的至少另一组dna分子与来自步骤(a)-(c)的至少三组消化的dna分子混合以产生至少单个归一化对照中包含四组dna分子,其中至少另一组dna分子是从样本中分离或衍生的。在某些实施方式中,使用本公开的方法从样本中分离或衍生出至少另一组dna分子。
在一些实施方式中,至少四组dna分子的预定量是相等的。
在一些实施方式中,至少四组dna分子的预定量不相等。在某些实施方式中,至少四组dna分子的预定量为线性序列或几何序列。
本公开内容提供了进行归一化对照的方法,包括:(a)合成至少一组dna分子,其中该组中的dna分子具有相同的序列,并且其中每个dna分子的序列包括5个以上的'至3',第一成分序列,第一限制性酶切位点,第二成分序列,第二限制位点和第三成分序列;(b)定量溶液中dna分子的数量;(c)用限制性酶消化至少一组dna分子,该酶切割第一和第二限制位点以产生至少一组消化的dna分子;(d)将预定量的消化的dna分亚组与预定量的至少两个另外的dna分亚组混合,以在单个归一化对照组合物中产生至少三个dna分亚组,其中从样本中分离或衍生出至少另外两组dna分子。
在一些实施方式中,至少三组dna分子的预定量是相等的。
在一些实施方式中,至少三组dna分子的预定量不相等。在一些实施方式中,至少三组dna分子的预定量为线性序列或几何序列。
在一些实施方式中,一组dna分子的预定量是基线,并且预定量的dna分子的其他组是基线的整数倍。在一些实施方式中,一组消化的dna分子的预定量是基线,并且其中每另外一组消化的dna分子的预定量是另一组消化的dna分子的预定量,不包括基线。在一些实施方式中,至少四组消化的dna分子的预定量的比例为1:2:4,任选地1:2:4:8或1:2:4:8:16等。
在一些实施方式中,样本包含质粒dna,线粒体dna,叶绿体dna或基因组dna。在某些实施方式中,样本包含从克隆载体分离的dna。在一些实施方式中,克隆载体是细菌组分染色体(bacterialcomponentchromosome,bac)、酵母人工染色体(yeastartificialchromosome,yac)、粘粒、福斯质粒或质粒。
样本制备
本公开提供了定量样本中至少一种靶核酸分子的表达水平的方法,其包括:(a)将已知量的本公开的归一化对照组合物与样本混合,(b)制备高通量测序文库,(c)对所述文库测序以产生读段集合,(d)将每个读段映射至样本或组合物,(e)确定由组合物中的多个核酸分子中的每个核酸分子产生的读段数目,(f)计算(a)中与样本混合的组合物中多个核酸分子各自的起始浓度与(c)中产生的读段数之间的关系,(g)建模(确定)该关系在读段和样本浓度之间,以及h。使用(g)中的模型,由靶核酸分子产生的读段的数目计算样本中至少一种靶核酸分子的初始浓度。
在一些实施方式中,样本包含核酸。核酸可以是dna,rna或dna和rna的混合物。核酸可以是单链的(例如,单链rna,单链的dna),双链的(例如,双链的dna,dna:rna杂交体)或其混合物。
在一些实施方式中,样本包含全血,血浆,血清,眼泪,唾液,粘液,脑脊液,牙齿,骨骼,指甲,粪便,尿液,组织或取自哺乳动物的活检物。在一些实施方式中,样本包括全血,血浆,血清,眼泪,唾液,粘液,脑脊液,牙齿,骨骼,指甲,粪便,尿液,组织或取自人的活检物。在一些实施方式中,样本包含来自植物的叶,花或其部分,茎,根,坚果或种子。在一些实施方式中,样本包含来自昆虫的身体部位,组织,粪便或血淋巴。
在一些实施方式中,该方法进一步包括从样本中提取核酸。在一些实施方式中,核酸包含dna,rna或其混合物。从样本中提取核酸的方法是本领域普通技术人员众所周知的。示例性的提取方法包括但不限于基于苯酚/氯仿的提取方法,以及诸如qiagenminiprep试剂盒(cat。no.27104)和qiagenrneasyminikit(cat。74104)。
在一些实施方式中,样本是哺乳动物样本。在一些实施方式中,靶核酸分子包含病毒,细菌,真菌或真核寄生虫的序列(例如,在哺乳动物样本中发现的病毒,细菌,真菌或真核寄生虫)。在一些实施方式中,靶核酸分子包含一种或多种哺乳动物基因。
在一些实施方式中,样本是人样本。在一些实施方式中,靶核酸分子包含病毒,细菌,真菌或真核寄生虫的序列(例如在人样本中发现的病毒,细菌,真菌或真核寄生虫)。在一些实施方式中,靶核酸分子包含一个或多个人类基因。
本公开内容提供了将已知量的本公开内容的归一化对照与样本混合的方法。在一些实施方式中,将归一化对照组合物与样本混合,使得归一化对照占从样本获得的总序列读段的一小部分。在一些实施方式中,归一化对照总体上应该代表使用ngs从样本获得的序列读段的一小部分(<5%)。
在一些实施方式中,相对于归一化对照中的多核苷酸的总量,归一化对照组合物中的多核苷酸的每个组或亚组的比率介于约0.001至约0.500:1之间。在一些实施方式中,相对于归一化对照中的多核苷酸的总量,归一化对照组合物中的多核苷酸的每个组或亚组的比率为约0.001至约0.420:1。在一些实施方式中,归一化对照组合物中的多核苷酸的每个组或亚组的比率介于约0.001至约0.450:1之间、介于约0.001至约0.400:1之间、介于约0.001至约0.400:1之间、介于约0.001至约0.350:1之间、介于约0.001至约0.300:1之间、介于约0.001至约0.250:1之间、介于约0.001至约0.200:1之间、介于约0.001至约0.150:1之间、介于约0.001至约0.100:1之间、介于约0.001至约0.05:1之间、介于约0.005至约0.500:1之间、介于约0.010至约0.500:1之间、介于约0.050至约0.500:1之间、介于约0.100至约0.500:1之间、介于约0.010至约0.400:1之间、介于约0.010至约0.300:1之间、介于约0.010至约0.200:1之间、介于约0.010至约0.100:1之间、介于约0.050至约0.400:1之间、介于约0.050至约0.300:1之间、介于约0.050至约0.200:1之间、或介于约0.050至约0.100:1之间。
在一些实施方式中,以介于约0.0001pg/μl与约45pg/μl之间、介于约0.001pg/μl与约45pg/μl之间、介于约0.001pg/μl与约10pg/μl之间、介于约0.005pg/μl与约5pg/μl或介于约0.009pg/μl与约5pg/μl之间的终浓度将组合物中的多核苷酸的每个组或亚组加入样本中。在一些实施方式中,以介于约0.009pg/μl与约5pg/μl之间的终浓度将组合物中的多核苷酸的每个组或亚组加入样本中。
在一些实施方式中,以介于约0.001阿摩尔/μl与约50阿摩尔/μl之间的终浓度将组合物中的多核苷酸的每个组或亚组加入样本中。在一些实施方式中,以介于约0.01阿摩尔/μl与50阿摩尔/μl之间的终浓度将组合物中的多核苷酸的每个组或亚组加入样本中。在一些实施方式中,以介于约0.05阿摩尔/μl与25阿摩尔/μl之间的终浓度将组合物中的多核苷酸的每个组或亚组加入样本中。
在一些实施方式中,该方法还包括从样本中提取核酸,并且在从样本中提取核酸之前进行归一化对照与样本的混合。在一些实施方式中,在核酸提取之前将归一化对照与样本混合允许归一化对照在样本制备期间测量核酸提取中的偏差。例如,与蛋白质复合的靶核酸可能对核酸提取有抵抗力,因此在ngs之后的阅读丰度中代表性不足。该过程可以通过添加适当的归一化对照来模仿,例如,被病毒衣壳蛋白封装的归一化对照。
在一些实施方式中,将归一化对照与样本混合在制备高通量测序文库之前进行。在一些实施方式中,特别是其中混合步骤在文库制备之前进行的那些实施方式中,nc测量测序样本中dna或rna靶序列向可测序文库分子的转化。与样本多核苷酸同时对nc多核苷酸的末端进行双索引可在文库制备过程中的任何时间跟踪每个样本转换的分子以及如何转换分子。这允许计算样本类型不可知库转换因子。这也允许将质量控制内置到样本制备中。使用此技术,可以比较试剂批次,酶促反应的效率以及许多其他有关测序的指标。这些比较可以按照测序反应的步骤进行,通常需要等到文库制备过程结束才能确定是否出现问题。
在一些实施方式中,将归一化对照与样本混合在与文库制备步骤同时发生。在库准备过程的任何时候都可以添加带有适当衔接子的归一化对照。
在一些实施方式中,在文库制备步骤之后发生归一化对照与样本的混合。具有适当的索引和/或测序衔接子的归一化对照可以在文库制备后但在ngs之前添加到文库中。在一些实施方式中,在该过程控制中在这一点上添加的归一化对照用于序列反应本身中的偏差。例如,具有高或低gc含量的归一化对照可以控制焦磷酸测序方法中已知的gc偏差。
文库
本公开的测序方法涉及测序文库的制备。测序文库的制备又涉及到一系列经衔接子修饰的dna片段集合的生产,所述集合已经准备好进行测序。多核苷酸的测序文库可以由dna或rna制备,包括dna或cdna的等同物,类似物,其是互补的或复制rna模板产生的dna,例如通过逆转录酶的作用。所述多核苷酸可以以双链dna(dsdna)形式(例如基因组dna片段、pcr和扩增产物)起源,或者多核苷酸可以以单链形式起源,如dna或rna,并已转换为dsdna形式。举例来说,可以将mrna分子复制到适合用于制备测序文库的双链cdna中。
用于某些ngs测序平台的测序文库的制备要求多核苷酸具有特定范围的片段大小,例如0-1200bp。因此,可能需要将大的多核苷酸片段化。无论多核苷酸是被强制片段化还是以片段形式自然存在,它们都将被转化为具有5-磷酸和3'-羟基的平末端dna。文库制备过程中的片段化过程是使用ngs定量靶序列时潜在的错误来源,可以通过在文库制备中的片段化步骤之前加入归一化对照来进行建模。
通常,使用本领域已知的方法或试剂盒对片段末端进行末端修复,例如平末端修饰。可通过酶处理,例如使用多核苷酸激酶,将平末端片段磷酸化。在一些实施方式中,例如通过某些类型的dna聚合酶(例如taq聚合酶或klenowexo减聚合酶)的活性将单个脱氧核苷酸(例如脱氧腺苷(a))添加至多核苷酸的3'末端。具有da尾的产物与在随后的步骤中连接至其的衔接子的每个双链体区域的3'末端上存在的't'突出端兼容。da尾部阻止了两个平末端多核苷酸的自连接,使得存在朝向衔接子连接的序列的形成的偏倚。将具有da尾的多核苷酸连接至双链衔接子多核苷酸序列。相同的衔接子可以用于多核苷酸的两个末端,或者可以使用两组衔接子。连接方法是本领域中已知的,并利用连接酶(例如dna连接酶)将衔接子共价连接至具有da尾的多核苷酸。衔接子可包含5'-磷酸部分以促进与靶3'-oh的连接。具有da尾的多核苷酸包含5'-磷酸部分,该部分是剪切过程中残留的,或者是使用酶处理步骤添加的,并已被末端修复,并任选地通过一个或多个突出的碱基进行延伸,以得到适用于连接的3'-oh。纯化连接反应的产物以去除未连接的衔接子、可能已经相互连接的衔接子,并选择用于簇产生的模板的大小范围,所述选择可以在扩增(例如pcr扩增)之前进行。连接产物的纯化可以通过包括凝胶电泳和固相可逆固定化(spri)的方法获得。
标准协议,例如使用例如illumina平台进行测序的协议,可以指示用户在da尾部之前纯化最终修复的产品,并在文库的衔接子连接步骤之前纯化da尾部产品。准备。末端修复产物和具有da尾的产物的纯化去除了酶、缓冲液、盐等,以为随后的酶促步骤提供有利的反应条件。在一些实施方式中,末端修复、da加尾和衔接子连接的步骤不包括纯化步骤。因此,在一些实施方式中,本公开的方法包括制备测序文库,所述制备测序文库包括末端修复、da加尾和衔接子连接的连续步骤。在制备不需要da加尾步骤的测序文库的实施方式(例如,使用roche454和solidtm3平台进行测序的方案)中,末端修复和衔接子连接的步骤不包括在衔接子连接之间进行末端修复产物的纯化步骤。
在一些实施方式中,制备扩增反应。扩增步骤将与流动池杂交所需的多核苷酸序列引入至衔接子连接的模板分子。扩增反应的内容物是本领域技术人员已知的,并且包括扩增反应所需的合适的底物(例如dntp),酶(例如dna聚合酶)和缓冲液组分。任选地,可以省略衔接子连接的多核苷酸的扩增。通常,扩增反应需要至少两个扩增引物,即引物寡核苷酸,它们可以是相同的,并且包括能够与待扩增的多核苷酸分子中的引物结合序列退火的“衔接子特异性部分”(如果互补,则可以互补)模板在退火步骤中被视为单链)。一旦形成,根据上述方法制备的模板文库可用于固相核酸扩增。如本文所用,术语“固相扩增”是指在固相支持物上或与固相支持物结合进行的任何核酸扩增反应,使得全部或部分扩增产物在形成时被固定在固相支持物上。特别地,该术语涵盖固相聚合酶链反应(固相pcr)和固相等温扩增,其类似于标准溶液相扩增的反应,除了将正向和反向扩增引物之一或两者固定在其上。坚实的支持。固相pcr涵盖了诸如乳液之类的系统,其中一种引物固定在珠粒上,另一种引物固定在游离溶液中,固相凝胶基质中的菌落形成,其中一种引物固定在表面,另一种引物固定在游离溶液中。扩增后,可通过微流体毛细管电泳分析测序文库,以确保文库中不含衔接子二聚体或单链dna。模板多核苷酸分子的文库特别适用于固相测序方法。除了提供用于固相测序和固相pcr的模板外,文库模板还提供了用于全基因组扩增的模板。
在一些实施方式中,使与衔接子连接的多核苷酸文库进行大规模平行测序,其包括用于对数百万个核酸片段进行测序的技术,例如,使用随机片段化的基因组dna附着于平面,光学透明的表面并进行固相扩增的技术。创建具有数百万个簇的高密度测序流动池。solexa/illumina方法依赖于随机片段化的基因组dna与平面光学透明表面的连接。延伸连接的dna片段并进行桥扩增,以创建具有数百万个簇的超高密度测序流动池,每个簇包含同一模板的数千个拷贝。使用强大的四色dna合成测序技术对簇模板进行测序,该技术采用可逆的终止子和可移动的荧光染料。或者,可以在珠粒上扩增文库,其中每个珠粒包含正向和反向扩增引物。
扩增文库的测序可以使用本文所述的任何合适的测序技术进行。
测序
本公开提供了使用下一代测序对样本进行测序的方法,该样本包含归一化对照。
在一些实施方式中,本文描述的方法采用下一代测序技术(ngs),其中克隆扩增的dna模板或单个dna分子在流动池中以大规模平行的方式进行测序(例如,如volkerding等人,clinchem55:641-658[2009];metzkermnaturerev11:31-46[2010]中所述)。除了高通量序列信息外,ngs还提供数字定量信息,因为每个读段的序列都是可计数的“序列标签”,代表单个克隆dna模板或单个dna分子。ngs的测序技术包括焦磷酸测序,使用可逆染料终止子的合成测序,通过寡核苷酸探针连接进行测序和实时测序。
测序技术中的一些测序技术是可商购的,例如来自affymetrixinc.(sunnyvale,calif.)的的杂交测序平台,以及来自454lifesciences(bradford,conn.)、illumina/solexa(hayward,calif.)和helicosbiosciences(cambridge,mass.)的合成测序平台,以及来自appliedbiosystems(fostercity,calif.)的连接测序平台。除了使用helicosbiosciences的合成测序进行的单分子测序外,本公开内容的方法还包括其他单分子测序技术,包括pacificbiosciences的smrttm技术,iontorrenttm技术和纳米孔测序例如,由牛津纳米孔技术公司开发。然而,本公开被设想为与任何未来开发的高通量测序技术兼容,该技术固有地具有低输入样本的定量问题。
虽然自动的sanger方法被认为是“第一代”技术,但是包括本公开的自动sanger测序在内的sanger测序也可以采用。
在本公开的方法的一些实施方式中,使用的dna测序技术是454测序(roche)(例如,如margulies,m等人,nature437:376-380[2005]中所述)。在454测序中,约300-800个碱基对的dna片段是平末端的,并将衔接子连接至所述片段的末端。衔接子用作片段的扩增和测序的引物。可以使用例如含有5'-生物素标签的衔接子b将片段附接到dna捕获珠,例如链霉亲和素包被的珠。在油水乳状液的液滴内pcr扩增与珠粒相连的片段。结果是有每个珠粒上克隆扩增的dna片段的多个拷贝。在第二步骤中,将珠粒捕获在孔(皮升大小)中。对每个dna片段并行进行焦磷酸测序。一个或多个核苷酸的添加产生光信号,该光信号由测序仪器中的ccd相机记录。信号强度与掺入的核苷酸数成比例。焦磷酸测序利用焦磷酸(ppi),所述焦磷酸在添加核苷酸后被释放。在腺苷5'磷酸硫酸盐存在下,ppi通过atp硫化酶转化为atp。萤光素酶使用atp将萤光素转化为氧化萤光素,该反应产生的光可以被识别和分析。454测序可产生长度介于100-1000bp之间的读段。
在本公开的方法的一些实施方式中,使用的dna测序技术是solidtm技术(appliedbiosystems)。在solidtm连接测序中,将5'和3'附接到dna片段以生成片段文库。接下来,在含有微珠,引物,模板和pcr成分的微反应器中制备克隆微珠种群。pcr之后,使模板变性,并富集珠粒以用延伸的模板分离珠粒。对所选珠粒上的模板进行3'修饰,使其可以结合到载玻片上。可以通过将部分随机的寡核苷酸与由特定荧光团鉴定的中心确定的碱基(或碱基对)进行顺序杂交和连接来确定序列。记录颜色后,将连接的寡核苷酸切割并去除,然后重复该过程。solid技术可以产生约50bp的读段。
在本公开的方法的一些实施方式中,使用的dna测序技术是pacificbiosciences的单分子实时(smrttm)测序技术。在smrt测序中,在dna合成过程中对染料标记核苷酸的连续掺入进行了成像。单个dna聚合酶分子连接到各个零模波长标识符(zmw标识符)的底表面,该标识符可在磷酸化核苷酸被掺入正在生长的引物链中时获得序列信息。zmw是一种限制结构,其使得能够在荧光核苷酸的背景下观察到dna核苷酸通过dna聚合酶掺入单个核苷酸,所述荧光核苷酸迅速扩散出zmw(以微秒为单位)。将核苷酸整合到一条正在生长的链中需要花费几毫秒的时间。在这段时间内,荧光标记被激发并产生荧光信号,并且荧光标记被切割掉。染料的对应荧光的鉴定表明掺入了哪个碱基。重复该过程。rs仪器的平均读段长度约为1100bp,有时读段的最大长度为2500或2900bp。rsii可以产生8,500-60,000bp的读段长度。
在本公开的方法的一些实施方式中,所使用的dna测序技术是纳米孔测序(例如,如在sonigv和mellera.clinchem53:1996-2001[2007]中所描述的)。纳米孔测序dna分析技术正在由包括牛津纳米孔技术公司(英国牛津)在内的许多公司进行工业开发。纳米孔测序是一种单分子测序技术,通过该技术,单分子dna在通过纳米孔时就可以直接测序。纳米孔是直径约为1纳米的小孔,纳米孔浸入导电流体中,由于离子通过纳米孔的传导,在纳米孔上施加电势(电压)会产生少量电流。流动的电流量对纳米孔的大小和形状敏感。当dna分子穿过纳米孔时,dna分子上的每个核苷酸都会以不同程度阻塞纳米孔,从而以不同程度改变通过纳米孔的电流大小。因此,当dna分子通过纳米孔时电流的这种变化代表了dna序列的读段。纳米孔minion可以产生超过10,000bp的读段长度。
在一些实施方式中,dna测序技术是离子激流单分子测序,其将半导体技术与简单的测序化学方法相结合,以将化学编码的信息(a、c、g、t)直接翻译成数字信息(0、1)。半导体芯片。实际上,当核苷酸通过聚合酶掺入dna链时,氢离子作为副产物释放出来。离子激流使用高密度的微型加工孔阵列以大规模并行方式执行此生化过程。每个孔中都保持不同的dna分子。孔下面是离子敏感层,在其下方是离子传感器。当将核苷酸(例如c)添加到dna模板中,然后整合到dna链中时,氢离子将被释放。来自该离子的电荷将改变溶液的ph值,可以通过iontorrent的离子传感器进行识别。测序仪实际上是世界上最小的固态ph计—调用碱基,直接从化学信息转变为数字信息。然后,离子个人基因组机(pgmtm)测序仪将一个核苷酸一个接一个地顺序注入芯片。如果下一个充满芯片的核苷酸不匹配。则将不记录任何电压变化,也不会调用任何碱基。如果dna链上有两个相同的碱基,则电压将加倍,并且芯片将记录两个相同的碱基被调用。直接鉴定允许在几秒钟内记录核苷酸掺入。离子激流可产生约400bp的读段长度。
在一些实施方式中,该方法采用illumina的基于合成的测序和基于可逆终止子的测序化学对数百万个dna片段进行大规模平行测序(例如,如bentley等,nature6:53-59[2009]中所述)。illumina的测序技术依赖于将片段化的基因组dna连接到一个平面的,光学透明的表面,该表面上结合了寡核苷酸锚。模板dna进行末端修复,以生成5'-磷酸化的平末端,而klenow片段的聚合酶活性用于将单个a碱基添加到平末端磷酸化dna片段的3'末端。该添加制备了用于与寡核苷酸衔接子连接的dna片段,该寡核苷酸衔接子在其3'端具有单个t碱基的突出端以增加连接效率。衔接子寡核苷酸与流动池锚互补。在有限稀释条件下,将衔接子修饰的单链模板dna添加到流通池中,并通过与锚点杂交的方法固定。延伸附着的dna片段并进行桥扩增,以创建具有数亿个簇的超高密度测序流动池,每个簇包含约1,000个相同模板的拷贝。使用强大的四色dna合成测序技术对模板进行测序,该技术采用可逆的终止子和可移动的荧光染料。使用激光激发和全内反射光学器件可以实现高灵敏度的荧光识别。将短序列读段与重复屏蔽的参考基因组进行比对,并使用专门开发的数据分析管道软件调用遗传差异。第一读段完成后,可以在原位重新生成模板,以从片段的相对端进行第二读段。因此,根据该方法使用dna片段的单端或双端测序。illumina的读段长度取决于特定的平台,但通常介于50-400bp之间。
定量
“式”、“算法”或“模型”是采用一个或多个连续或分类输入并计算输出值(有时称为“索引”或“索引值”)的任何数学方程式、算法、分析或编程过程或统计技术。“式”的非限制性示例包括总和、比率和回归算子(例如系数或指数)、生物标志物值的转换和归一化、统计分类模型,以及在历史群体上训练的神经网络。在确定样本中初始浓度与ngs之后的输出读段之间的关系时,线性和非线性方程式以及统计分类分析特别用于确定样本中初始浓度与ngs之后的序列读段之间的关系。
特别令人感兴趣的是利用模式识别功能的结构和句法统计分类算法,包括已建立的技术,例如互相关,主成分分析(pca),因子旋转,对数回归(logreg),线性判别分析(lda),支持向量机器(svm),随机森林(rf),递归分区树(rpart)以及其他相关的决策树分类技术,收缩质心(sc),stepaic,kth最近邻算法,boosting,决策树,神经网络,贝叶斯网络和隐马尔可夫模型等。这些可以与诸如akaike的信息标准(aic)或贝叶斯信息标准(bic)的信息标准结合,以便量化附加序列数据与模型改进之间的折衷,并有助于最小化过度拟合。生成的预测模型可以使用bootstrap,leave-one-out(loo)和10折交叉验证(10折cv)等技术在其他研究中验证,或在最初接受过培训的研究中交叉验证。在各个步骤,可以根据本领域中已知的技术通过值排列来评估错误发现率。
线性相关
在本公开的方法的一些实施方式中,归一化对照的组和/或亚组的浓度包含线性序列。在一些实施方式中,归一化对照的设计的线性允许库中读书之间的相关性。nc分子的浓度当加到样本中或在制备文库时完全相关并遵循线性模型。对于被这些这种nc多核苷酸共享的输出读段,这种关系仍然成立。当nc的初始浓度相对于对应归一化的读段计数绘制时,确定回归的r2值,r2大于0.8表示在没有任何错误的情况下遵循提取和下游文库制备过程。
将nc以一定的浓度和不同的大小加到库中。不同多核苷酸的大小范围意在表示要在文库中识别的靶dna或rna的插入物大小。由于预先确定在制备期间加到样本或文库中的nc的浓度,可以通过将靶的读段计数与通过nc确定的线性模型拟合来确定样本中存在的靶序列的浓度。在此模型中,在文库制备过程期间靶序列、样本中的背景核酸或nc的偏差最小。每个nc线性具有其自己的线方程,y=mx+c。y是给出的靶序列/nc的对数(每百万的读段,rpm);m是线的斜率并且c是截距。对于靶序列,x不确定。从理论上讲,nc遵循线性,可以计算为对数(浓度),以阿摩尔s/μl为单位。该浓度转换为每μl在文库中存在多少个靶序列片段。
这种模式的成功取决于r2大于0.8的nc的线性开始靶滴度评估。r2低于0.8表示在样本制备和/或高通量测序期间nc的质量控制失败。在该模型中,nc的线性基于绝对浓度建立。它进一步假设插入大小仅为一种,即存在100%基因组覆盖率和最小的样本变化。可以通过计算映射到背景样本序列(而非“靶”的样本序列)的读段百分比,映射到nc的读段百分比以及理论上的消耗百分比来确定样本变化。尽管需要靶序列的单个插入物大小,但这很少可以实现。理想地,在直方图中,选择靶插入物大小的3–5个最具代表性的箱,然后计算每个箱中的读段百分比。这样做本质上可以解决基因组覆盖问题。然后,插入分子的数量取决于这些箱中每个箱中的数量。
可以使用线性关系y=mx+c来计算靶插入分子的数量或浓度。靶分子数量(是以下的函数)滴度;和滴度f(插入大小箱,基因组覆盖率,%人映射,%nc映射)。
例如,为了进行质量控制,可以以彼此的比率添加归一化对照。这允许计算观察到的比率,并确认在整个过程/测定中核酸按预期进行转化。如果比率如预期的那样出来(例如,将nc1、nc2和nc3以1:2:4的浓度比率加到样本中,则读段计数/证据将恢复该比率-100个读段:200个读段:400个读段)。图1示出示例性归一化对照质量控制检查。例如,为了包含基因组dna而不是合成的dna多核苷酸例如噬菌体t4归一化nc的归一化对照的质量控制,寻找从该基因组(例如噬菌体t4基因组)中回收的最小读段数。
在一些情况下,使用nc对标准曲线进行归一化。在简单的情况下,要确定其丰度的观察读段(在本文中称为“靶”读段)除以观察到的nc读段(组合或单独读段,具体取决于哪种方式可以生成更好的标准曲线)。在这种情况下,归一化靶读段丰度如下计算:
归一化读段的丰度=[(观察到的靶)/(观察到的nc)]*c
(如果需要,其中c=常数)
图2显示在下一代dna文库测序中巨细胞病毒cmv(靶)病毒滴度与读段丰度的图,其中从向其添加cmvdna(靶)的血浆中提取dna。虚线表示cmv滴度对cmv读段丰度的线性回归。如果不进行归一化,则r2值为0.89。相反,在归一化的情况下,r2值为0.98(图3)。因此,归一化对照改善初始样本中的靶(此处为cmv)的丰度与文库制备和下一代测序后映射到靶的读段的百分比之间的相关性。
在文库制备之前将归一化对照加到样本中可以解释从影响样本中的核酸的不同实验条件和系统/随机误差出现的错误。当设计为尽可能接近实际生物学时,可以解释和归一化制备、转化、测序等方面的许多差异。
然而,在一些情况下,一些生物学因素无法模拟,并且因此无法被归一化对照解释。在这种情况下,原始读段到靶浓度的转换可以通过以下公式描述:
其中r是原始靶读段证据(或计数),f(r)是转换函数,该函数允许人们对所有所占变量的读段进行归一化,而δ函数是由于无法模拟或解释的过程而导致的残留误差。理想地,δ(r)变为零,并且该转换将原始靶读段证据转换为初始初始浓度(例如每毫升病毒拷贝)。简单函数f(r)的示例是简单的乘法因子。在这种情况下,f(r)由归一化对照的实际输入百分比/丰度和观察到的归一化对照的丰度(经由读段计数/证据)计算:
f(r)=f·r
其中f可以计算为
其他形式的f(r)可以解释并模拟靶性质,例如靶gc含量,靶大小,片段化条件,靶是否与蛋白质(例如衣壳中的病毒基因组)相关或为游离dna分子,等等。如果归一化对照旨在解释所有这些变量,则δ会越来越小。在图4的一个假设的示例中概述它的视觉表示。在图4中,使用下一代测序一式三份地分析从感染病毒的宿主分离的核酸样本。在图4中,每个下一代测序文库的最左边的条是起始样本中已知的非宿主浓度(病毒载量)(即样本中“靶”的起始丰度)。在文库制备后,下一代测序和读段比对后,对读段计数进行转化,以使它们恢复病毒载量。每个文库左侧第二个条是仅使用获得的病毒证据来计算病毒的丰度百分比的示例。图4证明,仅从获得的病毒证据计算起始样本中病毒的丰度百分比(病毒载量)时,每个文库之间存在巨大差异。该计算也不能反映原始样本中的实际病毒载量。使用f(r)函数,可以将原始读段证据转换为在文库之间更具可比性的量(图4,每个文库左起第三条)。使用f(r)函数,可以减少文库中计算出的病毒丰度百分比的可变性,并且所得的值更接近于实际的病毒载量。唯一直接影响病毒载量的剩余转化是δ(r)。准确计算δ(r)甚至可以进一步减少文库之间的可变性,并导致病毒载量(误差范围内)。
仔细构造归一化对照可以为每个文库和每个靶估算f(r)。可以使用随机序列,但也可以添加更多的生物学信息。这种方法的一个特点是,如果可以计算f(r),则可以避免运行多个样本,以将原始读段直接转换为初始样本中的靶丰度。f(r)缩小读段计数和靶丰度之间的差距,并降低可变性。可以通过设计实验来评估δ(r),该实验考虑到归一化对照中未包含的所有变量。例如,δ(r)可以归因于细胞壁,例如细菌细胞壁,或病毒的衣壳环境,其作用可以通过实验确定。通过用f(r)缩小初始样本中靶丰度与输出读段计数之间的差距,可以将δ最小化。可以从多个样本中计算可能是乘数因子(y=ax+b)的δ。在一些实施方式中,用于δ的高阶模型也可能是合适的。图5描绘使用f(r)而不是原始读段来计算病毒载量时预期的可变性降低。在运行许多样本以计算δr以获得图5中所示的线的方程式之前,将可变性通过f(r)尽可能减小。
f(r)和δr的计算后可变性的一个潜在来源是测定的执行位点。在一些情况下,可能是在次要位点执行测定时(例如,另一家医院计算患者样本中的病毒滴度),测定的执行递送并不完全落入针对该组靶和归一化对照最初生成的线的数据。在这种情况下,可以在次要位点验证该测定。例如,次要位点可以运行足够的样本来计算病毒载量转换,并将其方程式输入到web应用程序中,该web应用程序在根据实际样本计算病毒载量时使用。
影响样本到测序读段转化的一些可能变量的表在下文表示为表1。表1概述nc控制设计中要考虑的可能变量。表1概述当靶是病毒时在设计nc时要考虑的可能变量。然而,其中一些可能的变量可能适用于其他靶。
表1.nc设计中的变量
因此,本公开提供以下方法:计算与样本混合的归一化对照中多个核酸分子中的每个核酸分子的初始浓度与通过ngs产生的读段数目之间的关系,对读段与样本中浓度之间的关系进行建模,和使用该模型从通过靶核酸分子产生的读段的数目计算样本中的至少一个靶核酸分子的初始浓度。
在一些实施方式中,模型是线性模型。在一些实施方式中,样本中至少一种靶核酸分子的初始浓度与通过靶核酸分子产生的读段的数目的线性回归具有在归一化后大于0.95,大于0.96,大于0.97,大于0.98或大于0.99的r2值。在一些实施方式中,线性回归的r2值提高至少0.01、0.03、0.05、0.07、0.09、0.1、0.13、0.15、0.17、0.19、0.2、0.23、0.25、0.27、0.29、0.3、0.33、0.35、0.37、0.39、0.4、0.43、0.45、0.47、0.49、0.5、0.53、0.55、0.57、0.59、0.6、0.63、0.65、0.67、0.69、0.7、0.8、0.9、1.0或以下归一化之间的任何值。
使用机器学习确定滴度
支持向量机(svm)是一种监督学习方法的已知机制,以预测药物毒性和抗药性。它们是用于分类、回归和离群值检测的一组方法。为了使svm能够对数据进行预测,它必须与数据匹配。给定一组训练数据,标记为属于两个类别之一的每个数据点,svm训练算法建立一个将新数据分配到其中一个类别的模型。svm是使用监督的机器学习方法训练分类器的非概率二进制线性分类器。
在一些实施方式中,归一化对照数据用于拟合svm模型。在一些实施方式中,80%的数据用于训练svm,并且其余20%的数据用于测试svm。对模型的拟合充当反馈过程,其中仅考虑了线性模型。具有最接近进入ngs文库的归一化对照的实际浓度的预测的模型确定了用于预测样本中的靶序列滴度的模型。
因此,本公开提供以下方法:计算与样本混合的归一化对照中多个核酸分子中的每个核酸分子的初始浓度与通过ngs产生的读段数目之间的关系,和对读段和样本中的浓度之间的关系进行建模,和使用模型从靶核酸分子产生的读段数目计算样本中至少一个靶核酸分子的初始浓度。
在一些实施方式中,使用机器学习分类器创建模型。在一些实施方式中,监督机器学习分类器。在一些实施方式中,机器学习分类器是向量支持机器。
除非另有说明,否则所有百分比和比率均以分子的重量或数量(例如摩尔比)计算。适当的测量单位对于本领域的普通技术人员将是显而易见的。
除非另有说明,否则所有百分比和比率均基于总归一化对照组合物计算。
多分析物对照
本文提供多分析物对照、包括多分析物对照的组合物、制备多分析物对照的方法和使用多分析物对照的方法。如本文所述,多分析物对照包括已灭活的生物物种的混合物。在不限制可包含在多分析物对照中的生物物种的情况下,病毒、细菌、真菌和真核寄生虫(单细胞和多细胞二者)均被设想为可包含在多分析物对照中的混合物或物种中的生物物种。在一些实施方式中,多分析物对照还包含合适的载体、稀释剂或赋形剂。多分析物对照以及包含多分析物对照的组合物在本文中可互换地指代。
不希望受限于任何特定的应用,本公开的多分析物对照在高通量测序和样本分析中具有许多应用。在一些实施方式中,自主使用多分析物对照,即不使用本文所述的归一化对照。例如,在样本处理步骤(例如文库制备和测序)期间,可以将多分析物对照用作阳性对照。在一些实施方式中,多分析物对照与本文所述的归一化对照一起使用。例如,归一化对照可用于归一化来自多分析物对照中的一个或多个物种的读段。因此,在一些实施方式中,将归一化对照加到多分析物对照,其如本文所述进行处理和测序。由于预先确定多分析物对照中物质的浓度(滴度),多分析物对照可用作本文所述归一化对照的阳性对照。替代地或另外地,可以使用归一化对照来归一化多分析物对照中的一种或多种物种的读段,并且归一化的读段计数和滴度之间的关系用于生成校准曲线。该校准曲线可用于从读段计数中确定样本中靶生物的滴度,该读段计数也已使用本文所述的归一化对照进行归一化。在一些实施方式中,将归一化对照加到多分析物对照,并且对多分析物对照进行处理,测序并且与样本平行地归一化读段以产生校准曲线。在一些实施方式中,多分析物对照和归一化对照用于产生参考校准曲线。在一些实施方式中,将归一化对照和多分析物对照都加到样本中(即掺入)。在一些实施方式中,校准曲线用于计算样本中靶生物的滴度。
在一些实施方式中,本公开的归一化对照用于归一化来自多分析物对照的读段。在一些实施方式中,来自多分析物对照的归一化读段用于确定样本中的一种或多种靶生物的滴度。
本公开提供包含至少三种不同物种的生物的混合物的多分析物对照。在一些实施方式中,多分析物对照包括至少十种生物物种。在一些实施方式中,多分析物对照包含至少4种生物物种,至少5种生物物种,至少6种生物物种,至少7种生物物种,至少8种生物物种,至少9种生物物种,至少10种生物物种,至少11种生物物种,至少12种生物物种,至少13种生物物种,至少14种生物物种,至少15种生物物种,至少16种生物物种,至少18种生物物种,至少19种生物物种,至少20种生物物种,至少25种生物物种,至少30种生物物种,至少35种生物物种,至少40种生物物种,至少45种生物物种,至少50种生物物种,至少75种生物物种,至少100种生物物种,至少150种生物物种或至少200种生物物种。在一些实施方式中,多分析物对照基本上由10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28,29、30、31、32、33、34、35、36、37、38、39或40种生物物种组成。在一些实施方式中,多分析物对照基本上由10种生物物种组成。在一些实施方式中,多分析物对照基本上由11种生物物种组成。在一些实施方式中,多分析物对照中的生物已被灭活。在一些实施方式中,多分析物对照还包含可接受的载体、稀释剂或赋形剂。
在一些实施方式中,多分析物对照包含至少4种病毒,至少5种病毒,至少6种病毒,至少7种病毒,至少8种病毒,至少9种病毒,至少10种病毒,至少11种病毒,至少12种病毒,至少13种病毒,至少14种病毒,至少15种病毒,至少16种病毒,至少18种病毒,至少19种病毒,至少20种病毒,至少25种病毒,至少30种病毒,至少35种病毒,至少40种病毒,至少45种病毒,至少50种病毒,至少75种病毒,至少100种病毒,至少150种病毒或至少200种病毒。在一些实施方式中,多分析物对照基本上由10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28,29、30、31、32、33、34、35、36、37、38、39或40种病毒组成。在一些实施方式中,多分析物对照基本上由10种病毒组成。在一些实施方式中,多分析物对照基本上由11种病毒组成。在一些实施方式中,多分析物对照中的生物已被灭活。在一些实施方式中,多分析物对照还包含可接受的载体、稀释剂或赋形剂。
在本公开的多分析物对照的一些实施方式中,多分析物对照中的物种混合物中的每种物种以相同的滴度存在。
在一些实施方式中,每种物种以不同的滴度存在于多分析物对照中。在一些实施方式中,多分析物对照中的生物物种的滴度以预定的期望比率存在。如果预定的期望比率为1:1,则每种物种以相同的滴度存在。在替代示例中,在包含三种物种的多分析物对照中的每种生物可以以1:5:35或1:20:90的比率存在。设想在本公开的范围内的任何预定的期望滴度比。在一些实施方式中,在多分析物对照中的生物物种的滴度为线性序列、几何序列或对数序列。例如,在包含三种物种的多分析物对照中的物种可以以1:2:3或1:10:100的比率存在。本领域普通技术人员将能够选择适合于本文公开的多分析物对照的特定用途的预定期望比率。
在一些实施方式中,至少两种物种以相同的滴度存在于多分析物对照中。在一些实施方式中,例如其中多分析物对照包含至少三种物种的那些实施方式,至少两种物种以相同的滴度存在于多分析物对照中,而至少两种物种以不同的滴度存在。
在包含病原体物种的本公开的多分析物对照的一些实施方式中,混合物中的病原体的每种不同物种以每ml(iu/ml)约5至1000万个感染单位的浓度存在。在一些实施方式中,混合物中每种不同的物种以约50至500万iu/ml,约500至500万iu/ml,约500至100万iu/ml,约500至100,000iu/ml,约500至10,000iu/ml,或约1,000至10,000iu/ml的浓度存在。在一些实施方式中,每种物种是以0iu/ml,100iu/ml,1,000iu/ml,5,000iu/ml,10,000iu/ml,100,000iu/ml或1,000,000iu/ml存在的多分析物对照。
在一些实施方式中,以100iu/ml至1,000,000iu/ml,100iu/ml至100,000iu/ml,1,000iu/ml至1,000,000iu/ml,1,000iu/ml至100,000iu/ml,100iu/ml至10,000iu/ml或1,000iu/ml至10,000iu/ml的终浓度将多分析物对照加到样本中。在一些实施方式中,以100iu/ml、150iu/ml、1,000iu/ml、5,000iu/ml、10,000iu/ml或100,000iu/ml的终浓度将多分析物对照加到样本。在一些实施方式中,可以将多分析物对照以一定浓度,例如0iu/ml,100iu/ml,1,000iu/ml,5,000iu/ml,10,000iu/ml和100,000iu/ml范围加到多个样本。作为另一个示例,可以将多分析物对照以1iu/ml,100iu/ml,150iu/ml,1,000iu/ml,5,000iu/ml,10,000iu/ml和100,000iu/ml的浓度加到样本中。在一些实施方式中,该浓度范围用于计算用于确定样本中靶生物的滴度的校准曲线。
如本文所用,“滴度”或“物理滴度”是指溶液中生物的浓度。对于感染性生物,“滴度”或“感染滴度”是指每体积溶液的浓度感染单位(例如,感染单位或iu,每ml或μl)。测量滴度的方法对于本领域普通技术人员将是显而易见的。例如,可以通过测量溶液中生物的浓度来测量物理滴度。这可以使用直接方法(例如计数)或间接方法(例如通过测量特定于所讨论生物的蛋白质水平)来完成。物理滴度可以表示为每体积溶液中细胞,生物,颗粒或单位的数量(例如单位/ml)。测量感染滴度的方法是本领域普通技术人员已知的。例如,可以使用本领域常规的测定例如噬斑形成测定来确定病毒的感染滴度。在噬斑形成测定中,制备病毒原液的稀释液并铺在易感细胞单层上,并计数形成病毒噬菌斑的被感染细胞的数量。细菌滴度可以通过铺板并测量形成的菌落的数量,通过计数方法,分光光度法或通过本领域已知的任何其他方法来确定。
在本公开的多分析物对照的一些实施方式中,多分析物对照中的生物物种包括病毒,细菌,真菌,真核寄生虫或其组合的物种。
在一些实施方式中,在多分析物对照中的生物物种基本上由病毒物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150或至少200种病毒物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒物种组成。在一些实施方式中,在多分析物对照中的生物物种基本上由11种病毒物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种、至少4种、至少5种、至少6种、至少7种、至少8种、至少9种、至少10种、至少11种、至少12种、至少13种、至少14种、至少15种、至少20种、至少20种、至少25种、至少30种、至少35种、至少40种、至少45种、至少50种、至少75种、至少100种、至少150种或至少200种病毒物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒物种。在一些实施方式中,多分析物对照中的生物物种包括11种病毒物种。
在一些实施方式中,多分析物对照中的生物物种基本上由细菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种细菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种细菌物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150或至少200种细菌物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种细菌物种。
在一些实施方式中,在多分析物对照中的生物物种基本上由真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19种或20种真菌物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种真菌物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种真菌物种。
在一些实施方式中,多分析物对照中的生物物种基本上由病毒和细菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种病毒和细菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒和细菌物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种病毒和细菌物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒和细菌物种。
在一些实施方式中,在多分析物对照中的生物物种基本上由病毒和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种病毒和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种病毒和真菌物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒和真菌物种。
在一些实施方式中,在多分析物对照中的生物物种基本上由细菌和真菌组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种细菌和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种细菌和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种细菌和真菌物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种细菌和真菌物种。
在一些实施方式中,在多分析物对照中的生物物种基本上由病毒、细菌和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种病毒、细菌和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种基本上由3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒、细菌和真菌物种组成。在一些实施方式中,多分析物对照中的生物物种包括至少3种,至少4种,至少5种,至少6种,至少7种,至少8种,至少9种,至少10种,至少11种,至少12种,至少13种,至少14种,至少15种,至少20种,至少20种,至少25种,至少30种,至少35种,至少40种,至少45种,至少50种,至少75种,至少100种,至少150种或至少200种病毒、细菌和真菌物种。在一些实施方式中,多分析物对照中的生物物种包括3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20种病毒、细菌和真菌物种。
在一些实施方式中,多分析物对照中的生物物种包括一种或多种病毒物种。设想所有病毒物种都有资格包括在本公开的多分析物对照中。病毒可以是dna病毒、rna病毒或其组合。在一些实施方式中,该病毒是dna病毒或rna病毒。在一些实施方式中,dna病毒包含双链dna病毒基因组。在一些实施方式中,dna病毒包含单链dna病毒基因组。在一些实施方式中,rna病毒包含双链rna病毒基因组。在一些实施方式中,rna病毒包含单链rna病毒基因组。在一些实施方式中,单链rna病毒基因组包含有义链。在一些实施方式中,单链rna病毒基因组包含反义链。在一些实施方式中,病毒是通过baltimore分类系统确定的i型,ii型,iii型,iv型,v型,vi型或vii型病毒。
在一些实施方式中,多分析物对照包含作为人类病原体的病毒物种。在下表2中显示作为人类病原体的示例性但非限制性病毒:
表2.示例性人类病毒病原体
在一些实施方式中,多分析物对照中的生物物种包括一种或多种细菌物种。设想所有细菌物种都适合包含在本公开的多分析物对照中。在一些实施方式中,细菌是革兰氏阳性细菌或革兰氏阴性细菌。
在一些实施方式中,多分析物对照包括作为人类病原体的细菌物种。在下表3中显示作为人类病原体的示例性但非限制性细菌:
表3.示例性人类细菌病原体
在下表4中显示可包含在多分析物对照中的其他细菌物种,包括代表性的共生、共栖、致病性和抗药性物种,以及宏基因组学样本中发现的物种。
表4.示例性其他细菌物种
在一些实施方式中,多分析物对照包含一种或多种真核寄生虫。在一些实施方式中,该寄生虫是人寄生虫。在一些实施方式中,真核寄生虫是多细胞寄生虫(例如,圆虫物种)。在一些实施方式中,真核寄生虫是单细胞生物。在一些实施方式中,真核寄生虫是酵母或真菌。在一些实施方式中,真核寄生虫是未被分类为酵母或真菌(例如变形虫)的物种。
在一些实施方式中,多分析物对照包括一种或多种作为人类病原体的真菌。下表5显示作为人类病原体的示例性但非限制性真菌:
表5.示例性人类真菌病原体
可以包括在本公开的多分析物对照中的其他真菌物种包括但不限于新隐球菌和酿酒酵母。
在一些实施方式中,多分析物对照包括真核寄生虫物种。示例性但非限制性的真核寄生虫示于下表6中:
表6.示例性人类真核寄生虫
在示例性实施方式中,表7(改编自cdc的“应具报疾病的概述–美国,2019”,于2016年2月18日在wwwn.cdc.gov/nndss/conditions/notifiable/2019/)访问)提供进一步的示例性致病病原体,其可以使用本文提供的方法和组合物进行量化。
表7.示例性致病性病原体
在本公开的多分析物对照的一些实施方式中,多分析物对照中的生物的不同物种是人病原体。人类病原体的示例性物种包括表2-3和5-7中列出的任何物种。在多分析物对照中使用人类已知病原体允许多分析物对照对要使用本公开的方法确定其滴度的样本中的靶物种的行为进行建模。核酸提取的有效性,病原体基因组大小,文库制备效率,序列读段数和样本组成均会影响高通量测序文库的读段计数反映样本中靶生物的初始滴度的程度。包含与样本中的靶物种类似(例如,相同属)或相同物种的多分析物对照允许多分析物对照有效控制影响归一化读段反映样本中滴度的准确性的参数。在一些实施方式中,多分析物对照包括样本中的一种或多种靶生物。可以知道靶生物存在于样本中,或被怀疑存在于样本中(例如,多分析物对照包括一组常见的人类病原体)。
在一些实施方式中,人类病原体包括通常在血液或组织移植中使用的组织中发现的人类病原体。移植获得性感染是移植接受者的重要原因或发病率和死亡率。因此,本公开的多分析物对照可用作筛选一种或多种人类病原体的移植组织和血液中的阳性对照,从而降低被移植组织或血液感染的风险。通常在人类血液或组织移植物中发现的示例性病原体包括但不限于巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人类细小病毒b19(b19),人类免疫缺陷病毒(hiv),乙型肝炎,丙型肝炎,i型和ii型人类t淋巴细胞性病毒(htlv),西尼罗河病毒,寨卡病毒,链球菌种,金黄色葡萄球菌,粪肠球菌,蜡状芽孢杆菌,痤疮丙酸杆菌,粘质沙雷氏菌,沙雷氏菌粘菌,小肠结肠炎耶尔森菌,肠杆菌属,不动杆菌属,假单胞菌属,大肠杆菌,肺炎克雷伯菌,奇异变形杆菌,鲍氏不动杆菌,蜡状芽孢杆菌,凝固阴性葡萄球菌,链球菌,克雷伯菌属,沙雷氏菌属雷氏普罗威登斯菌,苍白密螺旋体,克鲁斯锥虫和小巴贝虫中的一种或多种。
在一些实施方式中,多分析物对照包含或基本上由11种病毒的混合物组成。示例性多分析物对照包括或基本上由以下组成:巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人细小病毒b19(b19)。在一些实施方式中,多分析物对照包括或基本上由10种病毒的混合物组成。示例性多分析物对照包括或基本上由以下组成:cmv(例如,菌株ad169),ebv,adv(例如,1型),bkv(例如,亚型1b-2),jcv(例如,1a型),hhv6a(例如,菌株gs),hhv6b(例如z-29菌株),hsv1(例如95菌株),hsv2(例如09菌株)和vzv(例如9/84菌株)。在一些实施方式中,多分析物对照包括或基本上由以下组成:cmv,ebv,adv,bkv,jcv,hhv6a,hhv6b,hsv1,hsv2和vzv。另一个示例性多分析物对照包括或基本上由以下组成:麻疹病毒,西尼罗河病毒,寨卡病毒,黄热病病毒,辛德比斯病毒,天花病毒,诺沃克病毒,狂犬病病毒和人鼻病毒。另一个示例性多分析物对照包括或基本上由以下组成:爱泼斯坦-巴尔病毒,人类巨细胞病毒,人类呼吸道合胞病毒,天花病毒,流感病毒,风疹病毒,腮腺炎病毒和人类sars冠状病毒。
可以构成本公开的多分析物对照的示例性生物混合物如下表8所示:
表8.示例性多分析物对照
在本公开的多分析物对照的一些实施方式中,多分析物对照中的生物的至少三个不同物种不是人类病原体。在一些实施方式中,多分析物对照中的至少三种不同物种的生物不是人类病原体,而是与作为人类病原体的物种处于相同属。相同属中的物种通常具有类似的物理特性,例如,相同属中的细菌可能具有类似的细胞壁,而病毒可能具有类似的病毒蛋白。当经历本文所述的样本处理方法时,可以期望相同属的物种表现类似。通过将多分析物对照中的物种与相同属的人类病原体(例如常见于移植组织或血液中的人类病原体)相匹配,多分析物对照可以模拟样本处理期间样本中的那些人病原性物种的行为。这可以产生本公开的方法的优异能力来计算样本中的一种或多种靶生物的滴度,例如使用本公开的多分析物对照和归一化对照的人临床样本的病原体。
在本公开的多分析物对照的一些实施方式中,多分析物对照中的物种包括人类病原体的物种和不是人类病原体的物种的混合物。
在一些实施方式中,多分析物对照还包括血浆。血浆可以来自任何脊椎动物。在一些实施方式中,血浆来自人,非人灵长类,小鼠,大鼠,兔,狗,猫,沙鼠,绵羊,猪,马,牛或驴。在一些实施方式中,血浆是人血浆。在一些实施方式中,血浆是合成产生的。
本文提供从本公开的任何一种多分析物对照中提取的核酸的集合。可以从本文所述的多分析物对照中提取核酸的方法对于本领域普通技术人员而言将是显而易见的。示例性方法包括但不限于苯酚/氯仿萃取,然后用乙醇或类似的合适溶剂沉淀,以及可商购的试剂盒,例如qiagen和zymomini-prep试剂盒。从多分析物对照中提取的核酸可用于本文所述的任何实施方式中。例如,从多分析物对照中提取的核酸可以在文库制备步骤中加到样本中,或与样本平行处理和测序,并用作阳性对照或生成校准曲线。
制备多分析物对照的方法
本公开提供制备本文所述的多分析物对照的方法。在一些实施方式中,该方法包括:(a)选择一组生物以包括在多分析物对照中;(b)在适当的培养条件下培养每种生物;(c)使每种生物灭活,和(d)将预定量的每种灭活生物与合适的载体、稀释剂或赋形剂混合以产生多分析物对照。
在多分析物对照中使生物失活的适当方法是本领域普通技术人员已知的。例如,许多病毒可以通过干热、蒸汽或低ph(例如小于4.0的ph)灭活。包膜病毒可以通过加入溶剂或去污剂使其失活,从而破坏病毒的包膜。也可以通过暴露于化学交联剂(例如甲醛)使病毒灭活。使细菌失活的方法包括紫外线,辐射,加热,低ph和用化学物质(例如环氧乙烷,甲醛和硫氰酸胍)处理。单细胞真核生物(例如变形虫或酵母)可以通过加热或化学交联剂(例如甲醛)进行灭活。
在一些实施方式中,生物已通过化学处理、热处理、ph处理或紫外线照射灭活。在一些实施方式中,化学处理包括甲醛。
在一些实施方式中,将多分析物对照中的已知量的灭活生物加到血浆中。血浆可以例如通过离心和/或过滤去除血细胞、血小板和其他细胞组分而与血液分离。血浆可以来自任何脊椎动物。在一些实施方式中,血浆来自人,非人灵长类,小鼠,大鼠,兔子,沙鼠,狗,猫,牛,绵羊,猪,马或驴。在一些实施方式中,血浆来自人。在样本处理期间,将灭活的生物加到血浆中可使多分析物对照紧密模拟实验样本的行为,例如包含人血浆的人临床样本。
使用多分析物对照的方法
本公开提供使用本文所述的多分析物对照的方法。在一些实施方式中,本公开的多分析物对照可以与样本,例如临床或实验样本并行处理。与样本并行处理的多分析物对照可用于产生校准曲线,本领域普通技术人员可使用该校准曲线确定包含生物混合物的样本中一种或多种生物的滴度。例如,样本可以是临床样本,其包含来自人宿主的细胞和来自非宿主例如病毒、细菌或真菌病原体的细胞或颗粒。在替代实施方式中,将多分析物对照直接加到样本,例如临床或实验样本,并用作内标以计算包含生物混合物的样本中一种或多种生物体的滴度。在一些实施方式中,多分析物对照用作本公开的方法的阳性对照。
在一些实施方式中,与样本平行但与样本分开处理多分析物对照,以产生高通量测序文库和来自其的读段的集合。在一些实施方式中,加到多分析物对照和样本的归一化对照用于归一化来自多分析物对照和样本的读段。在其中多分析物对照和样本被并行处理的那些实施方式中(即,没有将多分析物对照“掺入”样本中),多分析物对照包括作为人类病原体的物种、不是人类病原体的物种或其组合。在一些实施方式中,来自多分析物对照的归一化读段用于产生校准曲线,并且该校准曲线用于计算样本中靶生物的滴度。
在本公开的方法的一些实施方式中,所述方法用于量化多分析物对照中的每种生物物种的至少一个靶序列的表达水平。在一些实施方式中,所述方法包括(a)将已知量的归一化对照与多分析物对照混合,(b)制备高通量测序文库,(c)对所述文库测序以产生读段集合,(d)将来自读段集合的读段映射到多分析物对照或归一化对照,(e)确定被归一化对照中的多核苷酸的每个组或亚组产生的读段数量,(f)计算与(a)中的多分析物对照混合的归一化对照中的多核苷酸的每个组或亚组的初始浓度和(c)中产生的读段数之间的关系,(g)建模读段与多分析物对照中的每个物种的至少一个靶序列的浓度之间的关系,和(h)使用(g)中的模型通过靶序列产生的读段数,计算多分析物对照中的每个物种的至少一个靶序列的初始浓度。在一些实施方式中,该方法进一步包括从多分析物对照中提取核酸。在一些实施方式中,(a)的混合步骤在(b)的文库制备步骤之前,与(b)的文库制备步骤同时或在(b)的文库制备步骤之后进行。在一些实施方式中,(a)的混合步骤在从样本提取核酸之前进行。
在一些实施方式中,多分析物对照中的生物的每个不同物种包括至少一个靶序列。在一些实施方式中,至少一种靶序列是多分析物对照中的每种生物物种的不同靶序列。在一些实施方式中,多分析物对照中的每个物种包括一个或多个不同的靶序列。在一些实施方式中,在多分析物对照中来自每个生物物种的至少一个靶序列与来自多分析物对照中任何一种或多种其他生物的靶序列具有小于99%同一性,小于95%同一性,小于90%同一性,小于85%同一性,小于80%同一性,小于75%同一性,小于70%同一性,小于60%同一性,小于50%同一性,小于40%同一性,小于30%同一性或小于20%同一性。
在一些实施方式中,在多分析物对照中来自每个生物物种的至少一个靶序列与来自样本的靶序列相同,具有小于99%同一性,小于95%同一性,小于90%同一性,小于85%同一性,小于80%同一性,小于75%同一性,小于70%同一性,小于60%同一性,小于50%同一性,小于40%同一性,小于30%同一性,小于20%同一性,小于10%同一性或小于5%同一性。在一些实施方式中,在多分析物对照中来自生物物种的至少一个靶序列与来自样本的靶序列相同。在一些实施方式中,多分析物对照包括包含与来自样本的靶序列相同的靶序列的至少一种物种,和包含与来自样本的靶序列不相同的靶序列的至少一种物种。
在一些实施方式中,多分析物对照中的每种生物物种的至少一个靶序列包含基因组序列。在一些实施方式中,在多分析物对照中来自每个生物物种的至少一个靶序列包含该物种的参考基因组或由该物种的参考基因组组成。在一些实施方式中,在多分析物对照中来自每个生物物种的至少一个靶序列包含该物种的转录组或由该物种的转录组组成。在一些实施方式中,来自多分析物对照中的每个生物物种的至少一个靶序列包含该物种的参考基因组的一部分。所述至少一个靶序列可以是蛋白质编码序列或非编码序列。
在一些实施方式中,在步骤(d)将来自读段集合的读段映射到多分析物对照或归一化对照还包括将读段映射到多分析物对照(d)中的每个生物物种。来自高通量测序读段的集合的读段的映射方法对于本领域普通技术人员将是显而易见的。例如,在多分析物对照中的每个生物具有参考基因组的情况下,来自通过高通量序列生成的读段的集合的读段可以经由bwa,novoalign,bowtie,soap2,bfast,ssaha2,mpscan,gassst或perm算法映射到参考基因组。在一些实施方式中,将读段映射到多分析物对照中的每个生物的参考基因组包括将读段映射到多分析物对照中的每个生物物种的至少一个靶序列。
在一些实施方式中,在(h)计算至少一个靶序列的初始浓度包括在多分析物对照中为每种物种计算至少一个靶序列的初始浓度。多分析物对照中的每种物种的至少一种靶序列的初始浓度可以从读段计数与归一化对照中的每个组或亚组的初始浓度之间的关系计算,该关系使用归一化对照确定本公开的组合物和方法。
本公开提供量化样本中靶生物的滴度的方法。在一些实施方式中,所述方法包括(a)提供包含靶生物的样本,其中所述靶生物包含至少一个靶序列;(b)提供包含至少三种生物物种的已知滴度的多分析物对照,其中所述生物已被失活;(c)将已知量的本公开的归一化对照与样本和多分析物对照混合;(d)从样本和至少一个多分析物对照制备高通量测序文库;(e)对所述文库测序以产生样本读段集合和多分析物对照读段集合;(f)使用归一化对照使来自(e)的样本读段集合和多分析物对照读段集合归一化;(g)确定多分析物对照中归一化读段与已知生物物种滴度之间的关系;和(h)使用(g)中确定的关系计算样本中靶生物的滴度。
在一些实施方式中,生物物种各自包含至少一种物种特异性靶序列。在一些实施方式中,至少一种物种特异性靶序列在多分析物对照中的每个物种中是不同的。
在一些实施方式中,归一化来自多分析物对照的读段包括:(i)将来自样本读段集合的读段映射到样本或归一化对照;(ii)将来自多分析物对照读段集合的读段映射到多分析物对照或归一化对照;(iii)确定样本读段集合和多分析物对照读段集合的归一化对照中的多核苷酸的每一组或亚组产生的读段的数目;(iv)针对样本和多分析物对照,计算在归一化对照中的核苷酸的每个组或亚组的初始浓度和步骤(e)中产生的读段的数目之间的关系;以及(v)确定样本和多分析物对照中的读段与浓度之间的关系。
在一些实施方式中,确定多分析物对照和样本中的读段和滴度之间的关系包括:(i)从多分析物对照中的每个物种计算至少一种物种特异性靶序列的初始浓度;(ii)计算样本中的至少一个靶序列的初始浓度。在一些实施方式中,步骤(i)的映射进一步包括将样本读段集合映射到参考基因组。在一些实施方式中,参考基因组不包含靶序列。在一些实施方式中,参考基因组是人类基因组。例如,在包含人类宿主细胞和病原体,即包含靶序列的病原体的人类临床样本中,由样本的高通量测序文库产生的读段被映射到人类基因组作为参考基因组。映射到人类基因组并与人类基因组对齐的读段来自从人类细胞分离的核酸,并且不包含靶序列。未映射到人参考基因组的读段来自靶序列,并使用本公开的方法进行归一化。这种方法的优点是不一定需要提前知道产生靶序列的靶生物的身份。本公开的方法可以用于有效地确定样本中的未知靶生物的存在和滴度,例如人类临床样本中未知病原体。
在一些实施方式中,在步骤的映射进一步包括将样本读段集合映射到多于一个参考基因组。在一些实施方式中,第一参考基因组不包含靶序列,而第二参考基因组包含靶序列。例如,在包含人宿主细胞和病原体即包含靶序列的病原体的人临床样本中,将从样本的高通量测序文库产生的读段映射到不包含靶序列的人基因组,和对应于包含靶序列的病原体的第二参考基因组。在一些实施方式中,映射还包括针对多分析物对照中的每个生物物种,映射到物种特异性参考基因组。
在一些实施方式中,在多分析物对照中归一化读段与生物的已知滴度之间的关系是线性关系。在一些实施方式中,该关系是对数的。在多分析物对照中确定归一化读段与生物的已知滴度物种之间的关系的方法对于本领域普通技术人员而言将是显而易见的。
在一些实施方式中,多分析物对照用于产生校准曲线,该校准曲线用于从归一化读段计数确定样本中生物的滴度。示例性校准曲线在图13中显示并显示为相对于用于生成校准曲线的多分析物对照中生物的对数滴度的信号(归一化读段)。在一些实施方式中,校准曲线与一个或多个样本(例如临床或实验样本)平行产生。在一些实施方式中,校准曲线由在多种不同浓度下分析的多分析物对照产生。例如,可以从以1:10、1:100、1:1000等稀释的多分析物对照生成示例性校准曲线,然后进行测序。在一些实施方式中,校准曲线由以2个浓度,3个浓度,4个浓度,5个浓度,6个浓度,7个浓度,8个浓度,9个浓度或10个浓度提供的多分析物对照生成。在一些实施方式中,校准曲线包括参考校准曲线。例如,可以将样本中的靶序列的归一化读段与参考校准曲线进行比较,以确定样本中与靶序列相对应的生物的滴度。在一些实施方式,在例如采用参考校准曲线的那些实施方式中,与样本平行地处理一个或多个多分析物对照以验证校准曲线和/或用作阳性对照。例如,将高浓度和低浓度的两种多分析物对照与样本并行处理。
在本公开的方法的一些实施方式中,所述方法进一步包括将已知量的多分析物对照与样本混合(掺入方法)。多种分析物的对照用作内标以计算包含生物混合物的样本中的一种或多种生物的滴度。例如,样本可以是包含宿主细胞(人细胞)和一种或多种病毒颗粒或来自细菌或真菌病原体(非宿主)的细胞的人类临床样本。多分析物对照包括不是人类病原体的生物,因此可以使用序列差异将其与样本中的人类病原体区分开。
在一些实施方式中,多分析物对照包括与人类病原体相同属的生物。与人类病原体有关的生物在处理期间的行为将与样本中的人类病原体类似,因此为计算样本中的人类病原体的滴度提供优越的指标。在一些实施方式中,多分析物对照中的生物包含具有与人类病原体序列的小于99%同一性,小于95%同一性,小于90%同一性,小于85%同一性,小于80%同一性,小于75%同一性,小于70%同一性,小于60%同一性,小于50%同一性,小于40%同一性,小于30%同一性或小于20%同一性的靶序列。
制剂
本公开的归一化对照和多分析物对照组合物可以配制在本领域已知的任何可接受的载体、稀释剂或赋形剂中。
可以在溶液中以任何可接受的浓度、沉淀或冻干的组合物形式提供归一化对照。核酸例如归一化对照可以悬浮在溶液中,包括但不限于具有至多0.5mnacl的水,tris缓冲液,tris-edta缓冲液和tris-edta。
在本公开的制剂中使用的示例性缓冲剂包括三-(羟甲基)氨基甲烷(tris)的乙酸盐、硫酸盐、盐酸盐、磷酸盐或游离酸形式,尽管可以使用与tris相同的近似离子强度和pka的替代缓冲剂,其结果相同。除了缓冲盐之外,在组合物中还包括辅因子盐,例如钾(例如氯化钾或乙酸钾)和镁(例如氯化镁或乙酸镁)的那些。
可以将多分析物对照中的灭活生物体以溶液形式提供,冷冻干燥或冻干。如果冷冻干燥或冻干,可以将多分析物对照重新悬浮在适当体积的任何可接受的载体中。可接受的载体包括但不限于水,磷酸盐缓冲盐水(pbs)或林格氏溶液。
消耗或富集的方法
归一化对照、多分析物对照和使用它们的方法可以与富集感兴趣的靶序列的样本或消耗靶向耗尽的序列的样本的方法组合。在一些实施方式中,富集或消耗的方法包括基于核酸引导的核酸酶的方法,基于核苷酸修饰的方法或其组合。
因此,本公开提供量化或识别样本中的靶序列的方法,该方法包括消耗靶向消耗的序列的样本,加入本文所述的归一化对照和使用ngs对样本进行测序。可以在富集感兴趣的靶序列或靶向耗尽的核酸消耗之前、之中或之后加入归一化对照。
在一些实施方式中,在富集感兴趣的靶序列或靶向耗尽的序列消耗之前,将归一化对照加到测序样本中。在一些实施方式中,可以将归一化对照设计为模拟感兴趣的靶序列的那些特性,所述特性用于将那些序列与靶向耗尽的序列区分开。例如,如果使用核酸引导的核酸酶和靶向靶向耗尽的序列的多个引导核酸(gna)富集感兴趣的靶序列,则归一化对照不包含被多个gna靶向的序列。作为进一步的示例,如果使用基于核苷酸修饰的方法消耗靶向耗尽的序列,则归一化对照可以包括与富集的感兴趣的靶向序列的修饰类似的核苷酸修饰。
基于核苷酸修饰的方法
本文提供相对于靶向耗尽的核酸富集感兴趣的靶核酸的样本的方法,包括使用感兴趣的靶核酸与靶向耗尽的核酸之间核苷酸修饰的差异。在一些实施方式中,靶核酸包含测序样本中的靶序列,其存在和/或滴度将使用本公开的归一化对照和任选的多分析物对照来确定。可以在基于修饰的消耗方法之前将包含与感兴趣的靶序列相同的核苷酸修饰的归一化对照加到样本中。可替代地,可以在基于修饰的消耗后将归一化对照加到样本中。
设想在本公开的范围内的任何类型的核苷酸修饰。下文描述本公开的核苷酸修饰的示例性但非限制性示例。
由本公开的方法使用的核苷酸修饰可以在任何核苷酸(例如腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶或尿嘧啶)上发生。这些核苷酸修饰可发生在脱氧核糖核酸(dna)或核糖核酸(rna)上。这些核苷酸修饰可以发生在双链或单链dna分子上,或发生在双链或单链rna分子上。
在一些实施方式中,核苷酸修饰包括腺嘌呤修饰或胞嘧啶修饰。
在一些实施方式中,腺嘌呤修饰包括腺嘌呤甲基化。在一些实施方式中,腺嘌呤甲基化包括n6-甲基腺嘌呤(6ma)。在一些实施方式中,腺嘌呤甲基化包括由脱氧腺苷甲基化酶进行的dam甲基化。在一些实施方式中,腺嘌呤甲基化包括ecoki甲基化。在一些实施方式中,腺嘌呤修饰包括在n6处通过甘氨酸修饰的腺嘌呤(momylation)。
在一些实施方式中,修饰包括胞嘧啶修饰。在一些实施方式中,胞嘧啶修饰包括5-甲基胞嘧啶(5mc),5-羟基甲基胞嘧啶(5hmc),5-甲酰基胞嘧啶(5fc),5-羧基胞嘧啶(5cac),5-葡萄糖基羟甲基胞嘧啶(5ghmc)或3-甲基胞嘧啶(3mc)。在一些实施方式中,胞嘧啶甲基化包括5-甲基胞嘧啶(5mc)或n4-甲基胞嘧啶(4mc)。在一些实施方式中,胞嘧啶甲基化包括dcm甲基化,dnmt1甲基化,dnmt3a甲基化或dnmt3b甲基化。在一些实施方式中,胞嘧啶甲基化包括cpg甲基化、cpa甲基化、cpt甲基化、cpc甲基化,或它们的组合。在一些实施方式中,胞嘧啶甲基化包括cpg甲基化。例如,使用本公开的方法,cpg甲基化可以用于选择性地靶向哺乳动物基因组中的活性区域以进行消耗。
在一些实施方式中,胞嘧啶修饰包括5-羟甲基胞嘧啶(5hmc),5-甲酰基胞嘧啶(5fc)。5-甲酰基胞嘧啶是5mc,5-羧基胞嘧啶(5cac),5-葡萄糖基羟甲基胞嘧啶或3-甲基胞嘧啶的氧化衍生物。
在本公开的方法的一些实施方式中,所述方法至少使用第一修饰敏感的限制性酶和第二修饰敏感的限制性酶。在一些实施方式中,第一和第二修饰敏感的限制性酶是相同的。在一些实施方式中,第一和第二修饰敏感的限制性酶是不同的。在一些实施方式中,第一或第二修饰敏感的限制性酶是单一物种的限制性酶(例如,alui或mcrbc,但不是两者)。在一些实施方式中,第一或第二修饰敏感的限制性酶是2种或更多种修饰敏感的限制性酶的混合物(例如,fspei和abasi的混合物)。在本公开的方法的一些实施方式中,组合两种以上的不同方法,每种方法使用不同的修饰敏感的限制性酶或修饰敏感的限制性酶的混合物。
如本文所用,术语“修饰敏感的限制性酶”是指对限制性酶的识别位点内或附近的修饰核苷酸的存在敏感的限制性酶。可替代地或另外,修饰敏感的限制性酶可以对识别位点本身内的修饰核苷酸敏感。修饰敏感的限制性酶可以对与识别位点相邻的修饰核苷酸敏感,例如在识别位点的1-50个核苷酸,5'或3'内。本公开的核苷酸修饰可以在识别位点本身内,或包含与识别位点相邻的核苷酸(例如,在识别位点的1-50个核苷酸,5'或3',或两者内)。
能够阻断或降低修饰敏感的限制性酶的活性的示例性修饰包括但不限于n6-甲基腺嘌呤,5-甲基胞嘧啶(5mc),5-羟基甲基胞嘧啶(5hmc),5-甲酰基胞嘧啶(5fc),5-羧基胞嘧啶(5cac),5-葡萄糖基羟甲基胞嘧啶,3-甲基胞嘧啶(3mc),n4-甲基胞嘧啶(4mc)或其组合。能够阻断修饰敏感的限制性酶的示例性修饰包括由dam,dcm,ecoki,dnmt1,dnmt3a,dnmt3b和tet酶介导的修饰。
在一些实施方式中,修饰包括dam甲基化。被dam甲基化阻断的限制性酶包括但不限于alwi,bcgi,bcli,bsabi,bspdi,bspei,bsphi,clai,dpnii,hphi,hpy188i,hpy188iii,mboi,mboii,nrui,nt.alwi,taqαi和xbai。
在一些实施方式中,修饰包括dcm甲基化。被dcm甲基化阻断的限制性酶包括但不限于acc65i,alwni,apai,avai,avaii,bani,bsai,bsahi,bsli,bsmfi,bsski,bstxi,eaei,esp3i,ecoo109i,msci,nlaiv,pflmi,pspgi,pspomi,sau96i,scrfi,sexai,sfii,sfoi和stui。被cpg甲基化阻断的限制性酶包括但不限于aatii,accii,acii,acii,afei,agei,aor13hi,aor51hi,asci,asisi,alui,avai,bceai,bmgbi,bsai,bsahi,bsiei,bsiwi,bsmbi,bspdi,bspt104ibsrfalphai,bsshii,bstbi
bstui,cfr10i,clai,cpoi,eagi,esp3i,eco52i,faui,fsei,fspi,haeii,hgai,hhai,hpaii,hpych4iv,hpy99i,kasi,mlui,naei,ngomiv,noti,nrui,ntb。cvipii,nsbi,pmaci,psp1406i,pluti,pmli,pvui,rsriisacii,sali,smai,snabi,sfoi,sgrai,smai,srfi,sau3ai,tspmi和zrai。
在一些实施方式中,修饰敏感的限制性内切酶在包含至少一个修饰核苷酸的识别位点处是活性的,而在不包含至少一个修饰核苷酸的识别位点处不是活性的。在包含一个或多个修饰核苷酸的识别位点处裂解的修饰敏感的限制性酶识别的示例性修饰包括但不限于n6-甲基腺嘌呤,5-甲基胞嘧啶(5mc),5-羟基甲基胞嘧啶(5hmc),5-甲酰基胞嘧啶(5fc),5-羧基胞嘧啶(5cac),5-葡萄糖基羟甲基胞嘧啶,3-甲基胞嘧啶(3mc),n4-甲基胞嘧啶(4mc)或其组合。特异性裂解包含一个或多个修饰核苷酸的识别位点的示例性修饰识别的修饰敏感的限制性酶包括由dam,dcm,ecoki,dnmt1,dnmt3a,dnmt3b和tet酶介导的修饰。
在包含在识别位点内或附近的一个或多个修饰核苷酸的识别位点处裂解的示例性但非限制性的修饰敏感的限制性酶包括但不限于abasi,dpni,fspei,lpnpi,mspji和mcrbc。
在一些实施方式中,修饰包括5-葡糖基羟甲基胞嘧啶,并且修饰敏感的限制性酶包含abasi。abasi切割包含葡糖基羟甲基胞嘧啶的abasi识别位点,并且不切割不包含葡糖基羟甲基胞嘧啶的abasi识别位点。
在一些实施方式中,核苷酸修饰包含5-羟甲基胞嘧啶,且修饰敏感的限制性酶包含abasi和t4噬菌体β-葡萄糖基转移酶。t4噬菌体β-葡萄糖基转移酶可将尿苷二磷酸葡萄糖(udp-glc)的葡萄糖部分特异性转移到双链dna中的5-羟甲基胞嘧啶(5-hmc)残基,例如在abasi识别位点内,从而制备糖基羟甲基胞嘧啶修饰的abasi识别位点。abasi切割包含葡糖基羟甲基胞嘧啶的abasi识别位点,并且不切割不包含葡糖基羟甲基胞嘧啶的abasi识别位点。
在一些实施方式中,核苷酸修饰包含甲基胞嘧啶,而修饰敏感的限制性酶包含mcrbc。mcrbc切割包含甲基胞嘧啶的mcrbc位点,而不切割不包含甲基胞嘧啶的mcrbc位点。可以用一条或两条dna链上的甲基胞嘧啶修饰mcrbc位点。在一些实施方式中,mcrbc还切割一条或两条dna链上的包含羟甲基胞嘧啶的mcrbc位点。在一些实施方式中,mcrbc半位点被至多3,000个核苷酸隔开。在一些实施方式中,mcrbc半位点被55-103个核苷酸隔开。
在一些实施方式中,修饰包括腺嘌呤甲基化,并且所述方法包括用dpni消化。当gatc识别的两条链上的腺嘌呤被甲基化时,dpni会切割gatc识别位点。在一些实施方式中,同时包含腺嘌呤甲基化和胞嘧啶修饰的dpnigatc识别位点出现在细菌dna中,而不存在于哺乳动物dna中。包含甲基化腺嘌呤和修饰的胞嘧啶的这些识别位点可以通过样本中的dpni选择性切割(例如,混合的细菌和哺乳动物dna),然后用t4聚合酶处理以用未修饰的腺嘌呤和胞嘧啶替换切割末端的甲基化腺嘌呤和修饰的胞嘧啶。在存在模板、引物和核苷酸的情况下,t4聚合酶催化5'至3'方向的dna合成。t4聚合酶会将未修饰核苷酸整合到新合成的dna中。这产生现在在感兴趣的核酸中包含未修饰的胞嘧啶并且在靶向耗尽的核酸中包含修饰的胞嘧啶的样本。使用本公开的方法,修饰的胞嘧啶中的这些差异可用于富集感兴趣的核酸。
在本公开的方法的一些实施方式中,样本中的核酸被末端去磷酸化,从而使样本中的核酸与修饰敏感的限制性酶接触产生具有暴露的末端磷酸酯的感兴趣的核酸或靶向耗尽的核酸,这些暴露的末端磷酸酯可以在本公开的方法中使用以富集感兴趣的核酸的样本。例如,这些暴露的末端磷酸酯可用于靶向核酸被核酸外切酶降解而消耗(图28)或感兴趣的核酸用于衔接子连接(图27)。
如本文所用,术语“末端去磷酸化”是指具有从核酸分子的5'和3'末端去除的末端磷酸基团的核酸。在一些实施方式中,使用磷酸酶例如碱性磷酸酶将样本中的核酸末端去磷酸化。本公开的示例性磷酸酶包括但不限于虾碱性磷酸酶(sap),重组虾碱性磷酸酶(rsap),小牛肠碱性磷酸酶(cip)和南极磷酸酶。
如本文所用,术语“核酸外切酶”是指从核酸分子的3'或5'末端连续去除核苷酸的一类酶。核酸分子可以是dna或rna。dna或rna可以是单链或双链的。示例性核酸外切酶包括但不限于λ核酸酶,核酸外切酶i,核酸外切酶iii和bal-31。核酸外切酶可以用于使用本公开的方法选择性地降解靶向消耗的核酸(例如,图28)。
本公开提供与样本或感兴趣的核酸以及可选的归一化对照中的核酸的5'和3'末端连接的衔接子。在一些实施方式中,衔接子与归一化对照序列连接。在其他实施方式中,从头合成包含衔接子序列的归一化对照。
在本公开的方法的一些实施方式中,将衔接子连接到样本中的所有核酸,然后使用核苷酸修饰的差异来选择性地切割靶向耗尽的核酸,从而产生在两端被衔接子连接的感兴趣的核酸和在一端被衔接子连接的靶向耗尽的核酸(图29,图30)。在一些实施方式中,核苷酸修饰的差异用于选择性地消耗靶向耗尽的核酸,然后将衔接子连接到感兴趣的靶核酸(图28)。在一些实施方式中,核苷酸修饰的差异用于产生具有暴露的末端磷酸酯的感兴趣的核酸,所述末端磷酸酯用于将衔接子连接到感兴趣的靶核酸(图27)。
在本公开的方法的一些实施方式中,将衔接子连接到样本中的核酸的5'和3'末端。在一些实施方式中,衔接子还包括在5'末端和/或3'末端之间的插入序列。
在一些实施方式中,衔接子是可与双链dna分子的两条链连接的核酸。
在一些实施方式中,在消耗/富集之前连接衔接子。在其他实施方式中,衔接子在随后的步骤中被连接。
衔接子的非限制性示例包括线性,线性y形或发夹衔接子。在一些实施方式中,衔接子包含polyg序列。
在各种实施方式中,衔接子可以是发夹衔接子,即,一个与其自身碱基配对以形成具有双链茎和环的结构的分子,其中该分子的3'和5'末端分别连接到片段的双链dna分子的5'和3'末端。
可替代地,衔接子可以是连接到片段的一端或两端的y-衔接子,也称为通用衔接子。可替代地,衔接子本身可以由彼此碱基配对的两个不同的寡核苷酸分子组成。另外,衔接子的可连接末端可以设计成与通过限制性酶切割产生的突出端相容,或它可以具有平末端或5't突出端。在一些实施方式中,限制性酶是修饰敏感的限制性酶。
衔接子可以包括双链以及单链分子。因此,衔接子可以是dna或rna,或两者的混合物。含有rna的衔接子可以通过rnase处理或碱性水解来裂解。
衔接子的长度可以为10至100bp,尽管在不偏离本公开的情况下可以使用在该范围之外的衔接子。
衔接子可以配置用于下一代测序平台,例如,在illumina测序平台(例如hiseq或miseq)上使用,或在iontorrent平台上使用,或与nanopore技术一起使用。在一些实施方式中,衔接子包括测序衔接子(例如,illumina测序衔接子)。在一些实施方式中,衔接子包含独特的分子标识符(umi)序列,有时称为条形码。在一些实施方式中,umi序列包含对于每个原始核酸分子而言唯一的序列(例如,随机序列)。在一些实施方式中,衔接子包含多个不同的序列,例如每个核酸分子特有的umi,来自特定来源的核酸分子之间共享的条形码和测序衔接子。
靶向消耗的核酸可通过差异衔接子连接而消耗。在一些实施方式中,将衔接子连接到样本的核酸,并且随后基于其修饰状态从靶向消耗的核酸中去除一个或多个衔接子。例如,可以通过修饰敏感的限制性酶切割两端连接有衔接子的靶向消耗的核酸,从而产生在仅一端连接有衔接子的靶向消耗的核酸。后续步骤(例如,扩增)可用于仅靶向两端连接有衔接子的核酸,从而消耗靶向消耗的核酸。在另一个实施例中,样本的核酸被处理(例如,通过去磷酸化),使得仅被切割的核酸能够具有连接的衔接子;随后,感兴趣的核酸可以被修饰敏感的限制性酶(例如,从而暴露磷酸酯基团)切割,并且可以连接衔接子。后续步骤(例如,扩增)可用于仅靶向连接有衔接子的核酸,从而消耗靶向消耗的核酸。
靶向消耗的核酸可以通过消化(例如用核酸外切酶消化)来消耗。
靶向消耗的核酸可以通过大小选择来消耗。例如,修饰敏感的限制性内切酶可用于切割感兴趣的核酸或靶向耗尽的核酸,并且随后可基于由于切割导致的大小差异将感兴趣的核酸与靶向耗尽的核酸分离。
在一些情况下,不使用大小选择就消耗靶向消耗的核酸。
方案1:在图27中描述本文描述的本申请的示例性方法。对包含感兴趣的靶核酸(2701)和靶向耗尽的核酸(2702)以及可选的归一化对照的核酸样本进行末端去磷酸化(2705),以产生未磷酸化的感兴趣的核酸(2706)和靶向耗尽的核酸(2707)。在一些实施方式中,核酸在去磷酸化之前被片段化。在一些实施方式中,样本中的核酸被磷酸酶例如重组虾碱性磷酸酶(rsap)末端去磷酸化。在一些实施方式中,感兴趣的核酸和靶向耗尽的核酸都包括一个或多个用于修饰敏感的限制性酶(分别为2703,2704)的识别位点。在感兴趣的核酸中,修饰敏感的限制性内切酶的识别位点不包含修饰核苷酸(2703),或可替代地,比靶向耗尽的核酸的对应识别位点较不频繁地包含修饰核苷酸。在靶向耗尽的核酸中,修饰敏感的限制性酶的识别位点包含在限制位点内或附近的修饰核苷酸(2704),或可替代地,比感兴趣的核酸的对应识别位点更频繁地包含修饰核苷酸。修饰敏感的限制性酶(2709)的活性被其同源识别位点(2708)内或附近的修饰核苷酸的存在阻断,从而将修饰敏感的限制性酶的活性靶向到感兴趣的核酸(比较2710与2711)。在一些实施方式中,修饰敏感的限制性酶(2709)包括aatii,accii,aor13hi,aor51hi,bspt104i,bsshii,cfr10i,clai,cpoi,eco52i,haeii,hapii,hhai,mlui,naei,noti,nrui,nsbi,pmaci,psp1406i,pvui,sacii,sali,smai,snabi,alui或sau3ai。在一些实施方式中,修饰敏感的限制性酶(2709)包含alui或sau3ai。用修饰敏感的限制性内切酶消化样本(2713)产生在末端磷酸酯(2714)的5'和3'端具有末端磷酸酯的感兴趣的核酸。这些末端磷酸酯用于将衔接子(2715,连接步骤;116,衔接子)连接到感兴趣的核酸的末端,产生两端均衔接子连接的感兴趣的核酸(2717)。相反,靶向消耗的核酸没有衔接子连接(2711)。这些衔接子可用于下游应用,例如衔接子介导的pcr扩增,测序(例如高通量测序)和样本中靶核酸的量化和/或克隆。通过将衔接子与感兴趣的核酸连选择性地接,从而消耗靶向耗尽的核酸。无需使用大小选择即可完成该消耗。可替代地,将感兴趣的衔接子连接的核酸进行本文所述的一种或多种其他富集方法。例如,使衔接子连接的核酸经受本公开的另外的依赖修饰的富集方法(例如,图29中描绘的方法)。替代地或另外地,使衔接子连接的核酸经受本公开的基于核酸引导的核酸酶的富集方法(例如,图30中描绘的方法)。
方案2:在图28中描绘在此描述的本申请的示例性方法。对包含感兴趣的靶核酸(2801)和靶向耗尽的核酸(2802)以及任选的归一化对照的核酸的样本进行末端去磷酸化(2805)以产生未磷酸化的感兴趣的核酸(2806)和靶向耗尽的核酸(2807)。在一些实施方式中,核酸在去磷酸化之前被片段化。在一些实施方式中,样本中的核酸被磷酸酶例如重组虾碱性磷酸酶(rsap)末端去磷酸化。在一些实施方式中,感兴趣的核酸和靶向耗尽的核酸都包含一个或多个修饰敏感的限制性内切酶的识别位点(分别为2803和2804)。在感兴趣的核酸中,修饰敏感的限制性内切酶的识别位点不包含修饰核苷酸(2803),或可替代地,比靶向耗尽的核酸的对应识别位点较不频繁地包含修饰核苷酸。在靶向耗尽的核酸中,修饰敏感的限制性内切酶的识别位点包含在限制位点内或附近的修饰核苷酸(2804),或可替代地,比感兴趣的核酸的对应识别位点更频繁地包含修饰核苷酸。当在识别位点(2808)内或附近存在一个或多个修饰核苷酸时,修饰敏感的限制性内切酶(2809)切割其同源识别位点,而当识别位点不包含一个或多个修饰核苷酸(2808)时,则不切割其同源识别位点,从而将修饰敏感的限制性酶的活性靶向到靶向消耗的核酸(比较2810与2811)。在一些实施方式中,修饰敏感的限制性酶包括abasi,fspei,lpnpi,mspji或mcrbc。在一些实施方式中,修饰敏感的限制性内切酶是fspei。在一些实施方式中,修饰敏感的限制性内切酶是mspji。用修饰敏感的限制性内切酶(2812)消化样本产生在核酸的一端(2813)或5'和3'末端(2814)具有末端磷酸酯的靶向消耗的核酸。相反,未被修饰敏感的限制性酶切割的感兴趣的核酸在核酸的5'和/或3'末端没有暴露的末端磷酸酯(比较2810与2813-2814)。然后用核酸外切酶消化样本(2815,消化步骤;2816核酸外切酶),该酶使用靶向消耗的核酸中的末端磷酸酯以从核酸分子末端去除连续的核苷酸,从而从样本消耗靶向消耗的核酸。无需使用大小选择即可完成该消耗。在核酸外切酶消化后,将衔接子连接到感兴趣的核酸(2817),该核酸缺少末端磷酸酯,尚未被核酸外切酶消化。这产生在两端均衔接子连接的感兴趣的核酸(2818)。这些衔接子可用于下游应用,例如衔接子介导的pcr扩增,测序(例如高通量测序)和样本中靶核酸的量化和/或克隆。可替代地,将感兴趣的衔接子连接的核酸进行本文所述的一种或多种其他富集方法。例如,使衔接子连接的核酸经受本公开的另外的依赖修饰的富集方法(例如,图29中描绘的方法)。替代地或另外地,使衔接子连接的核酸经受本公开的基于核酸引导的核酸酶的富集方法(例如,图30中描绘的方法)。
方案3:在图29中描绘在此描述的本申请的示例性方法。将包含感兴趣的核酸(2901)和靶向耗尽的核酸(2902)以及任选地归一化对照的核酸样本进行衔接子连接(2905),或使其经受本公开的富集方法(2906)(例如,图27或图28中描绘的方法,其产生感兴趣的衔接子连接的核酸(2907)和靶向耗尽的衔接子连接的核酸(2908)。在一些实施方式中,感兴趣的核酸和靶向耗尽的核酸都包含一个或多个修饰敏感的限制性酶的识别位点(分别为2903和2904)。在感兴趣的核酸中,修饰敏感的限制性内切酶的识别位点不包含修饰核苷酸(2903),或可替代地,比靶向耗尽的核酸的对应识别位点较不频繁地包含修饰核苷酸。在靶向耗尽的核酸中,修饰敏感的限制性内切酶的识别位点在限制位点内或附近包含修饰核苷酸(2904),或可替代地,比感兴趣的核酸的对应识别位点更频繁地包含修饰核苷酸。当在识别位点(2908)内或附近存在一个或多个修饰核苷酸时,修饰敏感的限制性内切酶(2909)切割其同源识别位点,而当识别位点不包含一个或多个修饰核苷酸(2908)时,则不切割其同源识别位点,从而将修饰敏感的限制性酶的活性靶向到靶向消耗的核酸(比较2910与2911)。在一些实施方式中,修饰敏感的限制性酶包括abasi,fspei,lpnpi,mspji或mcrbc。在一些实施方式中,修饰敏感的限制性内切酶是fspei。在一些实施方式中,修饰敏感的限制性内切酶是mspji。样本用修饰敏感的限制性内切酶(2911)消化,产生未进行衔接子连接的靶向消耗的核酸(2912)或仅在一端进行衔接子连接(2913)。通过从靶向消耗的核酸中选择性地去除衔接子,从而消耗靶向消耗的核酸。无需使用大小选择即可完成该消耗。相反,未被修饰敏感的限制性酶切割的感兴趣的核酸在两端均被衔接子连接(对比2910与2912-2913)。这些衔接子可用于下游应用,例如衔接子介导的pcr扩增,测序(例如高通量测序)和样本中感兴趣的核酸的量化和/或克隆。
方案4:在图30中描述本文所述的示例性方法。多个gna(3001)用于将核酸引导的核酸酶(3002)靶向到衔接子连接的核酸样本中的靶向消耗的核酸(3003)。通过本文所述的任何富集方法均可产生衔接子连接的核酸,该方法在初始衔接子连接之前或之后,使用修饰敏感的限制性内切酶从样本中消耗靶向消耗的核酸。在这种方法中,gnas专门靶向到靶向耗尽的核酸(3003),而不是感兴趣的核酸(3004),因此不会被核酸引导的核酸酶(3002)切割。被核酸引导的核酸酶的切割导致靶向耗尽的核酸在一端被衔接子连接(3005),并且感兴趣的核酸在两端被衔接子连接(3003)。这些衔接子可用于下游应用,例如衔接子介导的pcr扩增,测序(例如高通量测序),样本中感兴趣的核酸的量化和克隆。
本文所述的任何方法均可用作独立方法以从样本中消耗靶向耗尽的核酸,从而富集靶核酸。可替代地,与单独的任何单个方法相比,本文所述的方法可以组合以获得更大程度的富集。
尽管本文描述方法的特定组合以及方法的组合的顺序,这些绝不旨在限制可以组合本公开的方法的方式。富集产生作为该方法的产物的感兴趣的衔接子连接的核酸的本公开的感兴趣的核酸的样本的任何方法可以与使用衔接子连接的核酸作为其起始底物的本公开的任何其他方法结合。
基于核酸引导的核酸酶的富集方法
本公开提供可与归一化对照和多分析物对照结合的用于富集靶序列的基于核酸引导的核酸酶的方法,以及任选地本文所述的其他富集或消耗方法。基于核酸引导的核酸酶的富集方法是采用核酸引导的核酸酶以富集感兴趣的序列的样本的方法。在wo/2016/100955、wo/2017/031360、wo/2017/100343、wo/2017/147345和wo/2018/227025中描述基于核酸引导的核酸酶的富集方法,其内容各自通过引用整体并入本文。
可以在对样本进行基于核酸引导的核酸酶的方法之前或之后,将归一化和任选的多分析物对照加到样本中。在一些实施方式中,归一化对照包括与本文所述的gna的靶向序列的序列不同的序列。
术语“核酸引导的核酸酶-gna复合物”是指包含核酸引导的核酸酶蛋白和引导核酸(gna,例如grna或gdna)的复合物。例如,“cas9-grna复合物”是指包含cas9蛋白和引导rna(grna)的复合物。核酸引导的核酸酶可以是任何类型的核酸引导的核酸酶,包括但不限于野生型核酸引导的核酸酶,催化死亡的核酸引导的核酸酶或核酸引导的核酸酶-切口酶。当核酸引导的核酸酶是crispr/cas核酸引导的核酸酶时,该复合物可以称为“crispr/cas系统蛋白-gna复合物”。
本公开的方法可以利用核酸引导的核酸酶。如本文所用,“核酸引导的核酸酶”是切割dna、rna或dna/rna杂交体并且使用一种或多种引导核酸(gna)赋予特异性的任何核酸酶。核酸引导的核酸酶包括crispr/cas系统蛋白以及非crispr/cas系统蛋白。本文提供的核酸引导的核酸酶可以是dna引导的dna核酸酶;例如,dna引导的rna核酸酶;rna引导的dna核酸酶;或rna引导的rna核酸酶。核酸酶可以是核酸内切酶。核酸酶可以是核酸外切酶。在一个实施方式中,核酸引导的核酸酶是核酸引导的dna核酸内切酶。在一个实施方式中,核酸引导的核酸酶是核酸引导的rna核酸内切酶。
在一些实施方式中,本公开的基于修饰的富集方法和基于核酸引导的核酸酶的富集方法消耗样本中的不同核酸,从而与单独的任一方法相比,实现所感兴趣的核酸更大程度的富集。
本文提供多个引导核酸(gna)(可互换地称为文库或集合)。在一些实施方式中,归一化对照不包含与多个gna中的任何gna的序列相同或高度类似的序列。
术语“引导核酸”是指能够与核酸引导的核酸酶和任选的其他核酸形成复合物的引导核酸(gna)。gna可以作为分离的核酸或作为核酸引导的核酸酶-gna复合物,例如cas9-grna复合物的一部分存在。
如本文所用,多个gna表示包含至少102个独特gna的gna的混合物。在一些实施方式中,多个gna包含至少102个独特gna,至少103个独特gna,至少104个独特gna,至少105个独特gna,至少106个独特gna,至少107个独特gna,至少108个独特gna,至少109个独特gna或至少1010个独特gna。在一些实施方式中,gna的集合包含总共至少102个独特gna,至少103个独特gna,至少104个独特gna或至少105个独特gna。
在一些实施方式中,gna的集合包含含有靶向序列的第一na片段;和包含核酸引导的核酸酶系统(例如,crispr/cas系统)蛋白结合序列的第二na片段。在一些实施方式中,第一和第二片段为5'至3'顺序。在一些实施方式中,第一和第二片段为3'至5'顺序。
在一些实施方式中,第一段的大小在多个gna中是12-100bp,12-75bp,12-50bp,12-30bp,12-25bp,12-22bp,12-20bp,12-18bp,12-16bp,14-250bp,14-100bp,14-75bp,14-50bp,14-30bp,14-25bp,14-22bp,14-20bp,14-18bp,14-17bp,14-16bp,15-250bp,15-100bp,15-75bp,15-50bp,15-30bp,15-25bp,15-22bp,15-20bp,15-18bp,15-17bp,15-16bp,16-250bp,16-100bp,16-75bp,16-50bp,16-30bp,16-25bp,16-22bp,16-20bp,16-18bp,16-17bp,17-250bp,17-100bp,17-75bp,或17-50bp,17-30bp,17-25bp,17-22bp,17-20bp,17-18bp,18-250bp,18-100bp,18-75bp,18-50bp,18-30bp,18-25bp,18-22bp,18-20bp,19-250bp,19-100bp,19-75bp,或19-50bp,19-30bp,19-25bp,或19-22bp。在一些特定的实施方式中,第一片段的大小是15bp,16bp,17bp,18bp,19bp或20bp。
在一些实施方式中,多个中的至少10%,或至少15%,或至少20%,或至少25%,或至少30%,或至少35%,或至少40%,或至少45%,或至少50%,或至少55%,或至少60%,或至少65%,或至少70%,或至少75%,或至少80%,或至少85%,或至少90%,或至少95%或100%的第一片段是15-50bp。
在一些实施方式中,多个gna包含靶向序列,所述靶向序列可与靶向消耗的核酸中的靶序列碱基配对,其中靶向消耗的核酸中的靶序列在样本中的靶向消耗的基因组或转录组中至少每1bp,至少每2bp,至少3bp,至少每4bp,至少每5bp,至少每6bp,至少每7bp,至少每8bp,至少每9bp,至少每10bp,至少每11bp,至少每12bp,至少每13bp,至少每14bp,至少每15bp,至少每16bp,至少每17bp,至少每18bp,至少每19bp,20bp,至少每25bp,至少每30bp,至少每40bp,至少每50bp,至少每100bp,至少每200bp,至少每300bp,至少每400bp,至少每500bp,至少每600bp,至少每700bp,至少每800bp,至少每900bp,至少每1000bp,至少每2500bp,至少每5000bp,至少每10,000bp,至少每15,000bp,至少每20,000bp,至少每25,000bp,至少每50,000bp,至少每100,000bp,至少每250,000bp,至少每500,000bp,至少每750,000bp,或甚至至少每1,000,000bp被间隔开。
在一些实施方式中,多个gnas包含含有靶向序列的第一na片段;和包含核酸引导的核酸酶系统(例如,crispr/cas系统)蛋白结合序列的第二na片段,其中多个中的gna可以具有多种第二na片段,这些片段对核酸引导的核酸酶系统(例如,crispr/cas系统)的蛋白质成员具有不同的特异性。例如,本文所提供gnas的集合可以包括其第二片段包含对第一核酸引导的核酸酶系统(例如,crispr/cas系统)蛋白特异的核酸引导的核酸酶系统(例如,crispr/cas系统)蛋白结合序列的成员;并且还包括其第二片段包含对第二核酸引导的核酸酶系统(例如,crispr/cas系统)蛋白特异的核酸引导的核酸酶系统(例如,crispr/cas系统)蛋白结合序列的成员,其中所述第一第二核酸引导的核酸酶系统蛋白(例如crispr/cas系统)并不相同。在一些实施方式中,本文提供的多个gna包含对cas9蛋白和选自以下的另一种蛋白表现出特异性的成员:cpf1,cas3,cas8a-c,cas10,casx,casy,cas13,cas14,cse1,csy1,csn2,cas4,csm2和cm5。包含靶向序列的第一na片段和包含核酸引导的核酸酶系统蛋白结合序列的第二na片段将取决于核酸引导的核酸酶系统蛋白。第一和第二na片段的合适的5'至3'排列以及核酸引导的核酸酶系统蛋白的选择对于本领域普通技术人员而言将是显而易见的。
在一些实施方式中,gna包含dna和rna。在一些实施方式中,gna由dna(gdna)组成。在一些实施方式中,gna由rna(grna)组成。
在一些实施方式中,gna包含grna,并且grna包含两个编码crrna和tracrrna的子段。在一些实施方式中,crrna不包含靶向序列加上可以与tracrrna杂交的额外序列。在一些实施方式中,crrna包含可以与tracrrna杂交的额外序列。在一些实施方式中,两个子段被独立地转录。在一些实施方式中,两个子段被转录为单个单元。在一些实施方式中,编码crrna的dna包含序列gttttagagctatgctgttttg(seqidno:1)的靶向序列5'。在一些实施方式中,编码tracrrna的dna包含序列ggaaccattcaaaacagcatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttt(seqidno:2)。
如本文所用,靶向序列是将gna引导至在样本中靶向消耗的核酸中的靶序列的靶向序列。例如,靶向序列靶向本文所述的任何非宿主序列。
本文提供包含片段的gna和多个gna,所述片段包含靶向序列。
在一些实施方式中,靶向序列包含dna或由dna组成。在一些实施方式中,靶向序列包含rna或由rna组成。
在一些实施方式中,靶向序列包含rna,并且与感兴趣的序列上的pam序列的序列5′共有至少70%序列同一性,至少75%序列同一性,至少80%序列同一性,至少85%序列同一性,至少90%序列同一性,至少95%序列同一性,或具有100%序列同一性,除了rna包含尿嘧啶而不是胸腺嘧啶外。在一些实施方式中,靶向序列包含rna,并且与感兴趣的序列上的pam序列的序列3'共有至少70%序列同一性,至少75%序列同一性,至少80%序列同一性,至少85%序列同一性,至少90%序列同一性,至少95%序列同一性,或共有100%序列同一性,除了rna包含尿嘧啶而不是胸腺嘧啶外。在一些实施方式中,pam序列是agg、cgg、tgg、ggg或nag。在一些实施方式中,pam序列是ttn、tcn或tgn。
在一些实施方式中,靶向序列包含rna并且与pam序列的核苷酸5'的核苷酸序列相反的链互补。在一些实施方式中,靶向序列与pam序列5'核苷酸序列相反的链至少70%互补,至少75%互补,至少80%互补,至少85%互补,至少90%互补,至少95%互补或100%互补。在一些实施方式中,靶向序列包含rna并且与pam序列的核苷酸3'的序列相反的链互补。在一些实施方式中,靶向序列与pam序列的核苷酸3'序列相反的链至少70%互补、至少75%互补、至少80%互补、至少85%互补、至少90%互补、至少95%互补或100%互补。在一些实施方式中,pam序列是agg、cgg、tgg、ggg或nag。在一些实施方式中,pam序列是ttn、tcn或tgn。
不同的crispr/cas系统蛋白识别不同的pam序列。pam序列可以位于靶向序列的5'或3'。例如,cas9可以识别位于靶向序列的紧邻3'末端的nggpam。cpf1可以识别位于靶向序列5'末端的ttnpam。被所有crispr/cas系统蛋白识别的所有pam序列被设想为在本公开的范围内。对于本领域普通技术人员而言,显而易见的是,pam序列与特定的crispr/cas系统蛋白相容。
本文提供gna和多个gna,其包含含有核酸引导的核酸酶蛋白质结合序列的片段。核酸引导的核酸酶可以是核酸引导的核酸酶系统蛋白(例如,crispr/cas系统)。核酸引导的核酸酶系统可以是rna引导的核酸酶系统。核酸引导的核酸酶系统可以是dna引导的核酸酶系统。
核酸引导的核酸酶蛋白质结合序列是与核酸引导的核酸酶系统的任何蛋白质成员结合的核酸序列。例如,crispr/cas蛋白质结合序列是结合crispr/cas系统的任何蛋白质成员的核酸序列。
在一些实施方式中,crispr/cas系统蛋白可以来自任何细菌或古细菌物种。在一些实施方式中,crispr/cas系统蛋白是分离的,重组产生的或合成的。在一些实施方式中,crispr/cas系统蛋白的示例可以是天然存在的或工程改造的版本。
在一些实施方式中,核酸引导的核酸酶系统蛋白(例如,crispr/cas系统蛋白)可以来自任何细菌或古细菌物种。在一些实施方式中,天然存在的crispr/cas系统蛋白可以属于casi类i,iii或iv型,或casii类ii或v型,并且可以包括cas9,cas3,cas8a-c,cas10,casx,casy,cas13,cas14,cse1,csy1,csn2,cas4,csm2,cmr5,csf1,c2c2,和cpf1。在一个示例性实施方式中,crispr/cas系统蛋白包含cas9。在一个示例性实施方式中,crispr/cas系统蛋白包含cpf1。
在一些实施方式中,核酸引导的核酸酶系统蛋白(例如,crispr/cas系统蛋白)来自或衍生自化脓性链球菌,金黄色葡萄球菌,脑膜炎奈瑟氏球菌,嗜热链球菌,树突密螺旋体,土拉弗朗西斯菌,多杀性巴斯德氏菌,空肠弯曲杆菌,海鸥弯曲杆菌,鸡败血支原体,nitratifractorsalsuginis,parvibaculumlavamentivorans,肠道罗斯拜瑞氏菌,灰质奈瑟氏球菌,重氮葡糖醋杆菌,固氮螺菌,sphaerochaetaglobus,柱状黄杆菌,流动型塔夫杆菌,嗜粪拟杆菌,运动支原体,法氏乳杆菌,巴氏链球菌,约氏乳杆菌,伪中间型葡萄球菌,filifactoralocis,嗜肺军团菌,suterellawadsworthensis和白喉杆菌,氨基酸球菌,毛螺科菌或普雷沃氏菌的核酸引导的核酸酶系统蛋白(例如,crispr/cas系统蛋白)。
在一些实施方式中,核酸引导的核酸酶系统蛋白结合序列包含gna(例如,grna)茎环序列。不同的crispr/cas系统蛋白与不同的核酸引导的核酸酶系统蛋白结合序列兼容。对于本领域普通技术人员而言,显而易见的是,crispr/cas系统蛋白与哪些核酸引导的核酸酶系统蛋白结合序列相容。
在一些实施方式中,编码gna(例如,grna)茎环序列的双链dna序列在一条链上包含以下dna序列(5'>3',gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttt)(seqidno:3),及其在另一链上的反向互补dna(5'>3',aaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctctaaaac)。
在一些实施方式中,编码gna(例如,grna)茎环序列的单链dna序列包含以下dna序列:(5'>3',aaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctctaaaac)(seqidno:4),其中单链dna充当转录模板。
在一些实施方式中,gna(例如,grna)茎环序列包含以下rna序列:(5'>3',guuuuagagcuagaaauagcgcguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcucuuuuuuuu)(seqidno:5)。
在一些实施方式中,编码gna(例如,grna)茎环序列的双链dna序列在一条链上包含以下dna序列(5'>3',gttttagagctatgctggaaacagcatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttttttc)(seqidno:6),及其在另一链上的反向互补dna(5'>3',gaaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatgctgtttccagcatagctctaaaac)。
在一些实施方式中,编码gna(例如,grna)茎环序列的单链dna序列包含以下dna序列:(5'>3',gaaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatgctgtttccagcatagctctaaaac)(seqidno:7),其中单链dna充当转录模板。
在一些实施方式中,gna(例如,grna)茎环序列包含以下rna序列:(5'>3',guuuuagagcuaugcuggaaacagcauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcgccgagucggugugcuuuuuuuc)(seqidno:8)。
在一些实施方式中,crispr/cas系统蛋白是cpf1蛋白。在一些实施方式中,所述cpf1蛋白分离或衍生自franciscella物种或acidaminococcus物种。在一些实施方式中,gna(例如,grna)crispr/cas系统蛋白结合序列包含以下rna序列:(5'>3',aauuucuacuguuguagau)(seqidno:9)。
在一些实施方式中,crispr/cas系统蛋白是cpf1蛋白。在一些实施方式中,所述cpf1蛋白分离或衍生自franciscella物种或acidaminococcus物种。在一些实施方式中,编码gna(例如,grna)crispr/cas系统蛋白结合序列的dna序列包含以下dna序列:(5'>3',aatttctactgttgtagat)(seqidno:10)。在一些实施方式中,dna是单链的。在一些实施方式中,dna是双链的。
在一些实施方式中,本文提供包含第一na片段和第二na片段的gna(例如,gna),所述第一na片段包含靶向序列,所述第二na片段包含核酸引导的核酸酶(例如crispr/cas)系统蛋白结合序列。在一些实施方式中,第一片段的大小是15bp,16bp,17bp,18bp,19bp或20bp。在一些实施方式中,第二片段包含单个片段,该单个片段包含grna茎环序列。在一些实施方式中,grna茎环序列包含以下rna序列:(5'>3',guuuuagagcuagaaauauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuuuuuu)(seqidno:5)。在一些实施方式中,grna茎环序列包含以下rna序列:(5'>3',guuuuagagcuaugcuggaaacagcauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcucuuuuuuuuc)(seqidno:8)。在一些实施方式中,第二片段包含两个子段:与第二rna子段(tracrrna)形成杂合体的第一rna子段(crrna),其共同作用以指导核酸引导的核酸酶(例如,crispr/cas)系统蛋白结合。在一些实施方式中,第二子段的序列包含guuuuagagcuaugcuguuuug。在一些实施方式中,第一rna片段和第二rna片段一起形成crrna序列。在一些实施方式中,将与第二rna片段形成杂交体的其他rna是tracrrna。在一些实施方式中,tracrrna包含5'>3'的序列,ggaaccauucaaaacagcauagcaaguuaaaauaagaguauagcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuuuuuu(seqidno:11)。
在一些实施方式中,本文提供包含第一na片段和第二na片段的gna(例如,gna),所述第一na片段包含靶向序列,所述第二na片段包含核酸引导的核酸酶(例如crispr/cas)系统蛋白结合序列。在一些实施方式中,例如其中crispr/cas系统蛋白是cpf1系统蛋白的那些实施方式,第二片段是第一片段的5'。在一些实施方式中,第一片段的大小是20bp。在一些实施方式中,第一片段的大小大于20bp。在一些实施方式中,第一片段的大小大于30bp。在一些实施方式中,第二片段包含单个片段,该单个片段包含grna茎环序列。在一些实施方式中,grna茎环序列包含以下rna序列:(5’>3’,aauuucuacuguuguagau)(seqidno:9)。
crispr/cas系统蛋白可以与野生型crispr/cas系统蛋白至少60%相同(例如,至少70%,至少80%或90%相同,至少95%相同或至少98%相同或至少99%相同)。crispr/cas系统蛋白可能具有野生型crispr/cas系统蛋白的所有功能,或只有一种或一些功能,包括结合活性和核酸酶活性。
术语“crispr/cas系统蛋白相关的引导na”是指引导na(gna)。crispr/cas系统蛋白相关的引导na可以作为分离的na存在,或作为crispr/cas系统蛋白-gna复合体的一部分存在。
在一些实施方式,crispr/cas系统蛋白是rna引导的dna核酸酶。在一些实施方式中,被crispr/cas系统蛋白切割的dna是双链的。切割双链dna的示例性rna引导的dna核酸酶包括但不限于cas9,cpf1,casx和casy。其他示例性rna引导的dna核酸酶包括cas10,csm2,csm3,csm4和csm5。在一些实施方式中,cas10,csm2,csm3,csm4和csm5与grna形成核糖核蛋白复合物。
在一些实施方式中,crispr/cas系统蛋白核酸引导的核酸酶是或包含cas9。本公开的cas9可以是分离的,重组产生的或合成的。在一些实施方式中,cas9蛋白是热稳定的。可在本文的实施方式中使用的cas9蛋白的一些示例可以在faran,l.cong,wxyan,dascott,jsgootenberg,ajkriz,b.zetsche,o.shalem,x.wu,ksmakarova,evkoonin,pasharp和f.zhang;“使用金黄色葡萄球菌cas9进行体内基因组编辑”,《自然》520,186–191(2015年4月9日)doi:10.1038/nature14299中找到,在此通过引用并入本文。在一些实施方式中,cas9是衍生自化脓性链球菌,金黄色葡萄球菌,脑膜炎奈瑟氏球菌,嗜热链球菌,树突密螺旋体,土拉弗朗西斯菌,多杀性巴斯德氏菌,空肠弯曲杆菌,海鸥弯曲杆菌,唾液分枝杆菌,唾液支原体,沙门氏菌,食管支原体,鸡败血支原体,nitratifractorsalsuginis,parvibaculumlavamentivorans,肠道罗斯拜瑞氏菌,灰质奈瑟氏球菌,重氮葡糖醋杆菌,固氮螺菌,sphaerochaetaglobus,柱状黄杆菌,流动型塔夫杆菌,嗜粪拟杆菌,运动支原体,法氏乳杆菌,巴氏链球菌,约氏乳杆菌,伪中间型葡萄球菌,filifactoralocis,嗜肺军团菌,suterellawadsworthensis和白喉杆菌的ii型crispr系统。
在一些实施方式中,cas9是衍生自化脓性链球菌的ii型crispr系统,并且pam序列是ngg,其位于靶特异性引导序列的紧邻3'末端。来自示例性细菌物种的ii型crispr系统的pam序列还可以包括:化脓性链球菌(ngg),金黄色葡萄球菌(nngrrt),脑膜炎奈瑟氏球菌(nnnngatt),嗜热链球菌(nnagaa)和树突密螺旋体(naaaac),这些在不脱离本公开的情况下都是可用的。
在一个示例性实施方式中,cas9序列可以例如从px330质粒(可得自addgene)获得,通过pcr再扩增,然后克隆至pet30(得自emdbiosciences)以在细菌中表达并纯化重组的6his标记的蛋白。
“cas9-gna复合物”是指包含cas9蛋白和引导na的复合物。cas9蛋白可以与野生型cas9蛋白例如化脓链球菌cas9蛋白至少60%相同(例如,至少70%,至少80%或90%相同,至少95%相同或至少98%相同或至少99%相同)。cas9蛋白可能具有野生型cas9蛋白的所有功能,或只有一种或一些功能,包括结合活性、核酸酶活性和核酸酶活性。
术语“cas9相关的引导的na”是指如上所述的引导的na。与cas9相关的引导的na可以单独存在,或作为cas9-gna复合体的一部分存在。
在一些实施方式中,crispr/cas系统蛋白核酸引导的核酸酶是或包含cpf1系统蛋白。本公开的cpf1系统蛋白可以是分离的、重组产生的或合成的。在一些实施方式中,cpf1蛋白是热稳定的。
cpf1系统蛋白是ii类、v型crispr系统蛋白。在一些实施方式中,cpf1蛋白分离或衍生自土拉弗朗西斯菌。在一些实施方式中,cpf1蛋白分离或衍生自酸氨基球菌、乳酸杆菌科细菌或普雷沃氏菌。
cpf1系统蛋白质结合到包含核酸引导的核酸酶系统蛋白-结合序列(例如,茎环)和靶向序列。cpf1靶向序列包含紧邻定位在靶核酸中的cpf1pam序列的3'的序列。与cas9不同,cpf1核酸引导的核酸酶系统蛋白-结合序列位于cpf1grna中靶向序列的5'。cpf1还可以在靶核酸中产生交错而不是平末端的切割。将cpf1蛋白-grna蛋白复合物靶向靶核酸后,弗朗西斯菌衍生的cpf1,例如以交错形式切割靶核酸离子,在靶向序列的3'端距pam18-23个碱基形成约5个核苷酸的5'突出端。相比之下,被野生型cas9切割在cas9pam的上游3个核苷酸处产生平末端。
示例性cpf1grna茎环序列包含以下rna序列:(5’>3’,aauuucuacuguuguagau)(seqidno:9)。
“cpf1蛋白-gna复合物”是指包含cpf1蛋白和引导na(例如,grna)的复合物。当gna是grna时,grna可以由单个分子,即与靶杂交并提供序列特异性的一个rna(“crrna”)组成。
cpf1蛋白与野生型cpf1蛋白可以具有至少60%同一性(例如,至少70%,至少80%或90%同一性,至少95%同一性或至少98%同一性或至少99%同一性)。cpf1蛋白可能具有野生型cpf1蛋白的所有功能,或只有一种或一些功能,包括结合活性和核酸酶活性。
cpf1系统蛋白可识别多种pam序列。被cpf1系统蛋白识别的示例性pam序列包括但不限于ttn,tcn和tgn。另外的cpf1pam序列包括但不限于tttn。cpf1pam序列的一个特征是,它们比被cas9蛋白使用的ngg或nagpam序列具有更高的a/t含量。
计算机系统和软件
本文描述的方法可以在计算机系统的上下文中使用,或用作存储在计算机可读存储介质中的软件或计算机可执行指令的一部分。
在一些实施方式中,系统(例如,计算机系统)可以用于实现本发明的一些实施方式的某些特征。
在一些实施方式中,该系统可以包括一个或多个存储器和/或存储设备。存储器和存储设备可以是一种或多种计算机可读存储介质,所述计算机可读存储介质可以存储实现本发明的各种实施方式的至少一部分的计算机可执行指令。
本文提供一种用于设计多个归一化对照多核苷酸序列的系统。在一些实施方式中,该系统包括计算机可读存储介质,该计算机可读存储介质存储计算机可执行指令,该计算机可执行指令包括:(i)用于导入至少一个参考序列的指令;(ii)从至少一个参考序列产生多个参考序列片段的指令;(iii)根据参考序列片段数目产生至少一个参数的分布的指令;(iv)将分布划分成箱的指令;(v)从箱的至少一个子集中选择多个参考序列片段的指令;和(vi)改组多个参考序列片段以产生改组序列的指令;从而产生多个归一化多核苷酸序列。在一些实施方式中,该系统包括计算机可读存储介质,该计算机可读存储介质存储计算机可执行指令,该计算机可执行指令包括:(i)用于导入至少一个参考序列的指令;(ii)从至少一个参考序列产生多个参考序列片段的指令;(iii)根据参考序列片段数目产生至少一个参数的分布的指令;(iv)将分布划分成箱的指令;(v)从箱的至少一个子集中的每一个中选择至少两个参考序列片段的指令,其中所述至少两个参考序列片段在参考序列中是不连续的,或来自不同的参考序列;(vi)连接来自每个箱的至少两个参考序列片段的指令;从而产生多个归一化多核苷酸序列。在一些实施方式中,该系统还包括处理器,该处理器被配置成执行包括以下步骤的步骤:(a)接收包括至少一个参考序列的一组输入文件;和(b)执行存储在计算机可读存储介质中的计算机可执行指令。在一些实施方式中,该参数包括以下各项中的至少一项:(1)gc含量百分比、(2)熵、(3)复杂性、(4)eiip、(5)长度,或它们的组合。
该计算系统可以包括一个或多个连接到互连的中央处理单元(“处理器”),存储器,输入/输出设备(例如键盘和指点设备),触摸设备,显示设备,存储设备(例如磁盘驱动器)和网络衔接子(例如网络接口)。互连是表示通过适当的桥、衔接子或控制器连接的任何一个或多个单独的物理总线、点对点连接或两者的抽象概念。因此,互连可以包括例如系统总线,外围组件互连(pci)总线或pci-express总线,hypertransport或行业标准结构(isa)总线,小型计算机系统接口(scsi)总线,通用串行总线(usb),iic(12c)总线或电气与电子工程师协会(ieee)标准1394总线,也称为firewire。
另外,数据结构和消息结构可以经由数据传输介质(例如,通信链路上的信号)来存储或传输。可以使用各种通信链路,例如internet、局域网、广域网或点对点拨号连接。因此,计算机可读介质可以包括计算机可读存储介质,例如非暂时性介质,以及计算机可读传输介质。
可以将存储在存储器中的指令实现为软件和/或固件以对一个或多个处理器进行编程以执行上述动作。在本发明的一些实施方式中,可以通过例如经由网络衔接子通过计算系统从远程系统下载这些软件或固件来将其最初提供给处理系统。
本文介绍的本发明的各种实施方式可以通过例如可编程电路(例如一个或多个微处理器)来实现,该可编程电路完全在专用的硬连线即非可编程电路中,或在这种形式的组合中用软件和/或固件进行编程。专用硬连线电路可以采用例如一个或多个asic、pld、fpga等形式。
可以根据算法来呈现详细描述的一些部分,该算法可以是对计算机存储器内的数据位的操作的符号表示。这些算法描述和表示是数据处理领域的技术人员用来最有效地将其工作的实质传达给本领域其他技术人员的那些方法。这里,算法通常被认为是导致所需结果的自洽操作序列。这些操作是需要对物理量进行物理操纵的操作。通常,尽管不是必须的,这些量采取能够被存储、传输、组合、比较和以其他方式操纵的电或磁信号的形式。主要出于通用的原因,有时已经证明将这些信号称为位,值,元件,符号,字符,项,数字等是方便的。
本文提出的算法和显示与任何特定的计算机或其他设备不是固有地相关。各种通用系统可以与根据本文的教导的程序一起使用,或可以证明构造更专用的装置以执行一些实施方式的方法是方便的。
此外,尽管已经在功能全面的计算机和计算机系统的上下文中描述实施方式,本领域技术人员将理解,各种实施方式能够以各种形式作为程序产品来分发,并且本公开同样适用,这与实际影响分发的机器或计算机可读介质的特定类型无关。
机器可读存储介质、机器可读介质或计算机可读(存储)介质的其他示例包括但不限于可记录类型的介质,例如易失性和非易失性存储设备,软盘和其他可移动磁盘,硬盘驱动器,光盘(例如光盘只读存储器(cdroms),数字通用磁盘,(dvd)等)以及传输类型的介质(例如,数字和模拟通信链路)。
试剂盒和制品
本文提供的试剂盒和制品包括本文所述的任何一种或多种归一化对照组合物和/或用于制备或使用本文所述的任何归一化对照组合物的试剂。在一些实施方式中,示例性试剂盒包含本文所述的dna归一化对照组合物。在一些实施方式中,示例性试剂盒包含本文所述的rna归一化对照组合物。在一些实施方式中,示例性试剂盒包含本文所述的dna和rna归一化对照组合物的组合。在一些实施方式中,试剂盒进一步包括使用归一化对照组合物的指令,例如稀释、浓度以及将归一化对照加到样本的指令。
在本文描述的试剂盒的一些实施方式中,试剂盒包含本公开的多分析物对照中的任何一种或多种,以及用于其使用的试剂。在一些实施方式中,该试剂盒还包括用于使用多分析物对照的指令,例如用于稀释、浓缩以及将多分析物对照加到样本,或与样本平行地处理多分析物对照的指令。本公开的试剂盒提供的示例性多分析物对照包括包含至少三种已经灭活的生物的多分析物对照。在一些实施方式中,试剂盒还包含用于稀释多分析物对照的试剂。在一些实施方式中,试剂盒进一步包括使用多分析物对照的指令。
在一些实施方式中,试剂盒包含归一化对照,多分析物对照以及试剂和使用它们的指令。在一些实施方式中,试剂盒进一步包含阳性对照。示例性阳性对照包括可以用于阳性对照或校准本文所述的归一化对照和多分析物对照的已知浓度的灭活生物(例如1iu/ml,10iu/ml,100iu/ml,1,000iu/ml,10,000iu/ml,100,000iu/ml或1,000,000iu/ml)。在一些实施方式中,阳性对照包括病毒,细菌,真菌,单细胞真核生物或其任何组合。
如本文所述,本文提供的试剂盒和制品包含任何一种或多种用于消耗靶向耗尽的序列的样本的试剂或用于在ngs测序之前富集感兴趣的序列的样本的试剂。在一些实施方式中,试剂盒包含任何一种或多种本文所述的组合物,不限于衔接子,gnas(例如,grnas或gdnas),gna集合(例如,grna或gdna的多个),改性敏感的限制性酶,对照和类似物。
在示例性实施方式中,试剂盒包含grna,其中grna靶向本文所述的任何宿主基因组或cdna序列。在进一步的示例性实施方式中,试剂盒包含grna,其中grna靶向人基因组或dna序列的其他来源。
如本文所述,本公开还提供用于进行使用核苷酸修饰的差异来富集感兴趣的核酸的样本的方法的所有必需试剂和指令。
本公开还提供试剂盒,所述试剂盒包含归一化对照,多分析物对照,用于消耗靶向耗尽的序列的样本的试剂或用于在ngs测序之前富集感兴趣的序列的样本的试剂,以及使用它们的试剂和指令。
本文还提供在使用本文提供的方法富集样本之前和之后监测信息的计算机软件。在一个示例性实施方式中,该软件可以在应用本文描述的方法之前和之后计算并报告样本中靶向耗尽的核酸序列的丰度,以评估脱靶消耗水平,并且其中该软件可以通过在使用本文提供的富集方法处理样本之前和之后比较感兴趣的序列的丰度,检查靶向耗尽/富集/捕获/分区/标记/调节/编辑的功效。
在本公开的试剂盒的一些实施方式中,试剂盒包含归一化对照,多分析物对照,用于样本富集和/或消耗的试剂,使用它们的指令,以及使用它们例如可接受的稀释剂的试剂。
在本公开的试剂盒的一些实施方式中,多分析物对照包含11种病毒物种的混合物或由11种病毒物种的混合物组成。在一些实施方式中,病毒由巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b)组成),1型单纯疱疹病毒(hsv1),2型单纯疱疹病毒(hsv2),水痘带状疱疹病毒(vzv)和人细小病毒b19(b19)组成。
在本公开的试剂盒的一些实施方式中,多分析物对照包含10种病毒物种的混合物或由10种病毒物种的混合物组成。在一些实施方式中,病毒由cmv,ebv,adv,bkv,jcv,hhv6a,hhv6b,hsv1,hsv2和vzv组成。
贯穿本公开给出的每个最大值限制包括每个较低的值限制,就像在本文中明确写出这些较低的值限制一样。贯穿本公开给出的每个最小值限制将包括每个更高的值限制,就像在本文中明确写出这些更高的值限制一样。贯穿本公开给出的每个数值范围将包括落入该较宽的数值范围内的每个较窄的数值范围,就好像这些较窄的数值范围均在本文中明确写出一样。
本文公开的值不应将其理解为严格限于所列举的确切数值。相反,除非另有说明,否则每个这样的值旨在表示所列举的值和围绕该值的功能上等效的范围。例如,公开为“20μm”的值旨在表示“约20μm”。
除非明确排除或以其他方式限制,否则本文引用的每个文件,包括任何交叉引用的或相关的专利或申请,均通过引用整体并入本文。引用任何文件并不意味着承认本文是本文公开或要求保护的任何发明的现有技术,或单独或与任何其他参考文献组合,教导,建议或公开任何这样的发明。此外,在本文档中术语的任何含义或定义与通过引用并入的文档中相同术语的任何含义或定义冲突的程度上,以在本文档中分配给该术语的含义或定义为准。
虽然已经图示和描述本公开的特定实施方式,在不脱离本公开的精神和范围的情况下,可以做出各种其他改变和修改。所附权利要求的范围包括在本公开的范围内的所有这样的改变和修改。
列举的实施方式
可以通过参考以下列举的说明性实施方式来定义本发明:
1.一种归一化对照,所述归一化对照包括至少三组多核苷酸,其中每个组内的多核苷酸具有相同的长度。
2.实施方式1的归一化对照,其中所述归一化对照中的所有多核苷酸的长度是相同的。
3.实施方式1的归一化对照,其中当与任何其他组内的多核苷酸相比时,每组中的多核苷酸具有不同的长度。
4.实施方式3的归一化对照,其中所述多核苷酸组的长度以线性序列或几何序列分布。
5.实施方式1-4中任一项的归一化对照,其中所述至少三个组中的多核苷酸的长度在约15bp至约50,000bp之间。
6.实施方式1-4中任一项的归一化对照,其中所述至少三个组中的多核苷酸的长度在约500bp至约1500bp之间。
7.实施方式1-4中任一项的归一化对照,其中所述至少三个组中的多核苷酸的长度在约100bp至约1200bp之间。
8.实施方式1-4中任一项的归一化对照,其中所述至少三个组中的多核苷酸的长度在约150bp至约600bp之间。
9.实施方式1-4中任一项的归一化对照,其中所述至少三个组包含具有选自以下的长度的多核苷酸:175bp,250bp和450bp;192bp,250bp和450bp;200bp,300bp和500bp;217bp,300bp和517bp;436bp,552bp和974bp;450bp,612bp和1034bp;510bp,626bp和1048bp;450bp,612bp和1034bp。
10.实施方式1-9中任一项的归一化对照,其中每个组内的多核苷酸包含相同的序列。
11.实施方式1-9中任一项的归一化对照,其中每个组内的多核苷酸不包含相同序列。
12.实施方式11的归一化对照,其中每组多核苷酸包含至少三个多核苷酸亚组,
其中每个亚组内的多核苷酸包含相同的序列,并且其中每个亚组内的多核苷酸不包含与任何其他亚组相同的序列。
13.实施方式10-12中任一项的归一化对照,其中所述至少一组多核苷酸中的至少一个序列包含组分序列。
14.实施方式10-12中任一项的归一化对照,其中每组多核苷酸中的每个序列均包含组分序列。
15.实施方式13或14的归一化对照,其中每组多核苷酸的组分序列与任何其他组的组分序列不同。
16.实施方式12的归一化对照,其中所述至少一组多核苷酸的至少一个亚组的序列包含组分序列。
17.实施方式12的归一化对照,其中所述至少一组的所述至少三个亚组中的每一个的序列包含组分序列。
18.实施方式12的归一化对照,其中每个组的每个亚组的序列包含组分序列。
19.实施方式17或18的归一化对照,其中每个亚组的组分序列与任何其他亚组的组分序列不同。
20.实施方式13-19中任一项的归一化对照,其中所述组分序列包含约6bp至约3000bp的序列。
21.实施方式13-20的归一化对照,其中所述组分序列包含随机序列。
22.实施方式10-13的归一化对照,其中所述至少一组多核苷酸的序列包含分离的序列。
23.实施方式10-12中任一项的归一化对照,其中每组多核苷酸的序列包含分离的序列。
24.实施方式12、13或15-17中任一项的归一化对照,其中所述至少一组多核苷酸的至少一个亚组的序列包含分离的序列。
25.实施方式12、13或15-17中任一项的归一化对照,其中所述至少一组的每个亚组的序列包含分离的序列。
26.实施方式12的归一化对照,其中每个组的每个亚组的序列包含分离的序列。
27.实施方式25或26中任一项的归一化对照,其中每个亚组的分离的序列与任何其他亚组的分离的序列不同。
28.实施方式22-26中任一项的归一化对照,其中所述分离的序列包含约6bp至约200,000bp,约15bp至约50,000bp,约500bp至约1500bp,约100bp至约1200bp,或约150bp至约600bp的序列。
29.根据实施方式22-28中任一项所述的归一化对照,其中所述分离的序列是分离的或来源于病毒、细菌、真菌或真核寄生虫。
30.实施方式29的归一化对照,其中所述病毒是t4噬菌体(t4)或巨细胞病毒(cmv)。
31.实施方式22-30中任一项的归一化对照,其中所述分离的序列是从质粒分离的。
32.实施方式22-31中任一项的归一化对照,其中所述分离的序列与测序样本中的至少一个靶序列不同。
33.实施方式1-32中任一项的归一化对照,其中归一化对照中的任何多核苷酸的序列具有小于或等于与测序样本中的至少一种靶序列的1%,2%,3%,4%,5%,6%,7%,8%,9%,10%,11%,12%,13%,14%,15%,16%,17%,18%,19%,20%,30%,40%,50%、60%,70%,80%,85%,90%,95%,96%,97%,98%或99%同一性。
34.实施方式12-33中任一项的归一化对照,其中所述归一化对照中的多核苷酸的至少一个亚组的至少一个序列与所述测序样本中的所述至少一个靶序列具有至少一种序列特性。
35.实施方式34的归一化对照,其中所述至少一个序列特性包括gc含量百分比,熵,复杂性,长度,电子-离子相互作用电势(eiip),转座元件序列的序列特性,病毒序列的序列特性,细菌序列的序列特性,真菌序列的序列特性,真核寄生虫序列,一个或多个人类基因序列的序列特性或其任何组合。
36.实施方式35的归一化对照,其中病毒序列的特性包括gc含量百分比,重复序列,反向末端重复(itr)序列,内部核糖体进入位点(ires),蛋白质编码序列,转录后调控元件序列,转录调控元件,启动子序列,顺式作用rna元件,rna结构元件的序列,基因组包装信号,5'非翻译区(5'utr)序列,3'非翻译区域(3'utr)序列或其组合。
37.实施方式35的归一化对照,其中细菌序列的特性包括gc含量百分比,重复序列,microrna结合位点,内部核糖体进入位点(ires),蛋白质编码序列,转录调控元件序列,启动子序列,5'utr序列,3'utr序列或其组合。
38.实施方式37的归一化对照,其中所述重复序列元件包含polya基序,polyt基序,polyg基序,polyc基序,二核苷酸基序,三核苷酸基序,四核苷酸基序,五核苷酸基序,六核苷酸基序,七核苷酸基序,八核苷酸基序,九核苷酸基序,散布的重复序列元件,核糖体rna序列,trna序列或其组合。
39.实施方式35的归一化对照,其中所述真菌序列的特性包括gc含量百分比,重复序列,microrna结合位点,内部核糖体进入位点(ires)序列,蛋白质编码序列,转录调节元件,启动子序列,5'utr序列,3'utr序列,着丝粒序列,端粒序列,亚端粒序列,线粒体序列或其组合。
40.实施方式35的归一化对照,其中一个或多个人类基因的特性包括gc含量百分比,重复序列,蛋白质编码序列,内含子序列,5'utr序列,3'utr序列,转录调控元件,启动子序列,microrna结合位点或其组合。
41.实施方式34-40中任一项的归一化对照,其中测序样本包含宿主和非宿主核酸的混合物。
42.实施方式41的归一化对照,其中所述宿主是真核生物。
43.实施方式41的归一化对照,其中所述宿主是昆虫,植物或动物。
44.实施方式43的归一化对照,其中所述动物是人。
45.实施方式41-44中任一项的归一化对照,其中所述非宿主包括共生体,共生生物,寄生虫或病原体。
46.实施方式45的归一化对照,其中所述非宿主包括多种物种。
47.实施方式41-44中任一项的归一化对照,其中所述非宿主包括病毒物种,细菌物种,真菌物种,真核寄生虫物种或其组合。
48.实施方式32-47中任一项的归一化对照,其中所述至少一个靶序列是测序样本中的非宿主序列。
49.实施方式48的归一化对照,其中所述非宿主是病毒,细菌或真菌,并且所述至少一个靶序列包含毒力因子序列。
50.实施方式49的归一化对照,其中所述毒力因子包含一种或多种基因编码内毒素,外毒素,溶血素,蛋白酶,脂肪酶,dna酶,粘附素,侵袭素,抗吞噬剂,破坏性酶或gtp酶活性的调节剂。
51.实施方式48的归一化对照,其中所述非宿主是细菌,并且所述至少一个靶序列包含抗生素抗性基因的序列。
52.实施方式32-51中任一项的归一化对照,其中所述至少一个靶序列包含多个参考序列。
53.实施方式52的归一化对照,其中多个参考序列包含至少2个、至少10个、至少50个、至少100个、至少200个、至少300个、至少400个、至少500个、至少600个、至少700个、至少800个、至少900个、至少1,000个、至少1,200个、至少1,300个、至少1,400个、至少1,500个、至少1,600个、至少1,700个、至少1,800个、至少1,900个、至少2,000个、至少2,200个、至少2,400个、至少2,600个、至少2,800个、至少3,000个、至少4,000个、至少5,000个、至少6,000个、至少7,000个、至少8,000个、至少9,000个或至少10,000个参考序列。
54.实施方式53的归一化对照,其中所述参考序列包含非宿主序列。
55.实施方式53或54的归一化对照,其中所述参考序列包含病毒序列,细菌序列,真菌序列或其组合。
56.实施方式52-55中任一项的方法,其中所述多个参考序列包含来自多种生物的参考序列。
57.实施方式56的归一化对照,其中所述参考序列包含基因组dna序列或cdna序列或其组合。
58.实施方式52-57中任一项的归一化对照,其中所述分离的序列包含至少一个已经改组的参考序列的至少一个片段。
59.实施方式58的归一化对照,其中所述分离的序列包含至少第一参考序列和第二参考序列的至少两个片段,所述至少两个片段已经被改组和连接。
60.实施方式58的归一化对照,其中所述第一参考序列和第二参考序列在生物的基因组中不相邻。
61.实施方式58的归一化对照,其中第一和第二参考序列来自不同的生物。
62.实施方式58-61中任一项的归一化对照,其中所述参考序列的至少一个片段包含参考序列的约10-100,约10-80,约20-60,约20-40,约20-30或约20-25个连续核苷酸。
63.实施方式52-62中任一项的归一化对照,其中:
a.所述至少一个参考序列由参考序列片段的分布表示;
b.分布划分为至少5个箱;和
c.多个参考序列片段选自至少5个箱中的至少3个,将其改组并任选地连接,从而产生分离的序列。
64.实施方式63的归一化对照,其中所述参考序列片段相对于参考序列长度和(1)gc含量百分比、(2)熵、(3)复杂性、(4)eiip、(5)长度或其组合中的至少一个而分布。
65.实施方式63或64的归一化对照,其中每个箱代表所述分布的1%,2%,5%,10%,15%或20%。
66.实施方式63或64的归一化对照,其中每个箱代表gc含量分布的5%,10%,15%或20%。
67.实施方式63-66中任一项的归一化对照,其中归一化对照的每组内的多核苷酸通过改组和任选地连接选自至少3个箱之一的参考序列片段而产生。
68.实施方式65-67中任一项的归一化对照,其中每个组中的多核苷酸具有以下中的至少三者:(1)与参考序列分布中对应箱的参考序列片段类似的gc含量百分比,(2)类似的熵,(3)类似的eiip,(4)类似的长度,(5)类似的复杂性。
69.实施方式63-68中的任一项的归一化对照,其中参考序列片段选自gc含量分布百分比的第10,第30,第50,第70,第80和第90个百分位箱。
70.实施方式52-57中任一项的归一化对照,其中所述分离的序列包含连接的至少一个参考序列的至少两个片段,并且其中所述至少两个片段在所述至少一个参考序列中不相邻。
71.实施方式52-57中任一项的归一化对照,其中所述分离的序列包含至少第一参考序列的至少第一片段和至少第二参考序列的至少第二片段,其中所述至少第一和第二片段是连接的,并且其中第一和第二参考序列不是相同的参考序列。
72.实施方式70或71的归一化对照,其中所述至少第一和第二片段是对应的参考序列的约15-60,约20-40,约20-30,约15-32、20-32或约25-35个连续核苷酸。
73.实施方式70或71的归一化对照,其中所述至少第一和第二片段是对应参考序列的29、30、31、32、33或34个连续核苷酸。
74.实施方式70-73中任一项的归一化对照,其中所述至少第一和第二片段小于测序读段的平均长度的一半。
75.实施方式70-74中任一项的归一化对照,其中:
a.至少一个参考序列由使用滑动窗口产生的重叠参考序列片段的分布表示;
b.分布划分为至少5个箱;和
c.从至少5个箱中的至少3个中选择多个参考序列片段并连接,从而产生分离的序列。
76.实施方式75的归一化对照,其中所述参考序列片段相对于参考序列长度和(1)gc含量百分比、(2)熵、(3)复杂性、(4)eiip或其组合中的至少一个而分布。
77.实施方式75或76的归一化对照,其中每个箱代表分布的1%,2%,5%,10%,15%或20%。
78.实施方式75或76的归一化对照,其中每个箱代表gc含量分布的5%,10%,15%或20%。
79.实施方式75-78中任一项的归一化对照,其中每个组内的多核苷酸通过连接选自至少3个箱之一的参考序列片段而产生。
80.实施方式75-79中任一项的归一化对照,其中每个组中的多核苷酸具有以下至少三个:(1)与参考序列分布中的对应箱的参考序列片段类似的gc含量百分比,(2)类似的熵,(3)类似的eiip,(4)类似的长度,(5)类似的复杂性。
81.实施方式75-80中任一项的归一化对照,其中所述滑动窗包括1bp,2bp,3bp,4bp或5bp的滑动窗。
82.实施方式75-81中的任一项的归一化对照,其中参考序列片段选自gc含量分布百分比的第10,第30,第50,第70,第80和第90个百分位箱。
83.实施方式32-80中任一项的归一化对照,其中所述测序样本包括来自被诊断患有癌症的受试者的样本。
84.实施方式83的归一化对照,其中所述癌症包括细胞群体,所述细胞群体包含相对于所述受试者的非癌性细胞或来自健康受试者的非癌性细胞具有至少一个遗传改变的序列。
85.实施方式84的归一化对照,其中所述至少一个遗传改变包括单核苷酸多态性(snp),插入,缺失,倒位或染色体重排。
86.实施方式83或84的归一化,其中所述靶序列包含至少一种遗传改变。
87.实施方式32-82中任一项的归一化对照,其中所述测序样本包括宏基因组样本。
88.实施方式87的归一化对照,其中所述宏基因组样本是环境样本。
89.实施方式88的归一化对照,其中所述至少一个靶序列包含宏基因组学样本内的物种的序列。
90.实施方式88的归一化对照,其中所述至少一个靶序列包含毒力因子,抗生素抗性基因的序列或与致病性相关的序列。
91.实施方式1-90中任一项的归一化对照,其中每个组的多核苷酸处于相同浓度。
92.实施方式1-90中任一项的归一化对照,其中每组的多核苷酸不具有相同的浓度。
93.实施方式92的归一化对照,其中所有组的多核苷酸处于线性序列或几何序列的浓度。
94.实施方式93的归一化对照,其中一组多核苷酸具有基线浓度,并且其中剩余的非基线组的浓度是基线浓度的整数倍。
95.实施方式93的归一化对照,其中一组多核苷酸具有基线浓度,并且其中每组多核苷酸的浓度是归一化对照中的另一组的浓度的两倍,不包括基线浓度。
96.实施方式93的归一化对照,其中所述至少三组多核苷酸以1:2:4的浓度比率存在。
97.实施方式1-96中任一项的归一化对照,其中组内的多核苷酸具有相同的gc含量百分比。
98.实施方式97的归一化对照,其中组内的多核苷酸与每个其他组内的多核苷酸具有相同的gc含量百分比。
99.实施方式97的归一化对照,其中组内的多核苷酸与任何其他组内的多核苷酸具有不同的gc含量百分比。
100.实施方式97-99中任一项的归一化对照,其中每个组内的多核苷酸的gc含量百分比在40%至60%之间,包括端点。
101.实施方式97-99中任一项的归一化对照,其中每个组内的多核苷酸的gc含量百分比在43%至56%之间,包括端点。
102.实施方式1-101中任一项的归一化对照,其中所述多核苷酸包含编码唯一分子标识符的序列。
103.实施方式102的归一化对照,其中编码唯一分子标识符的序列包含第一索引序列或第二索引序列。
104.实施方式1-103中任一项的归一化对照,其中所述多核苷酸包含脱氧核糖核酸(dna)分子,核糖核酸(rna)分子或dna-rna杂合分子。
105.实施方式104的归一化对照,其中多核苷酸在归一化对照中的浓度在每微升(μl)约0.005至约500皮克(pg)之间。
106.实施方式105的归一化对照,其中所述多核苷酸在归一化对照中的浓度为约0.5至约50pg/μl。
107.实施方式105的归一化对照,其中所述多核苷酸在归一化对照中的浓度为1.4pg/μl。
108.实施方式1-107中任一项的归一化对照,所述归一化对照还包含至少一种蛋白质。
109.实施方式108的归一化对照,其中所述至少一种蛋白质分离或衍生自细菌,病毒或真核寄生虫。
110.实施方式108的归一化对照,其中所述至少一种蛋白质是病毒衣壳蛋白或细菌细胞壁蛋白。
111.实施方式108-110中任一项的归一化对照,其中所述多核苷酸群和所述蛋白质可操作地连接。
112.一种制备归一化对照的方法,所述方法包括:
a.合成至少三组dna分子,其中一组内的dna分子具有相同的序列,并且其中每个dna分子的序列在5'至3'之间包含第一组分序列,第一限制性酶切位点,第二组分序列,第二限制性位点和第三组分序列;
b.对溶液中所述多组dna分子中的每个组的量进行定量;
c.用切割第一和第二限制位点的限制性酶消化所述多组dna分子中的每个组,以产生至少三组经消化的dna分子;以及
d.将预定量的所述多组经消化的dna分子中的各个组混合,从而产生归一化对照。
113.实施方式112的方法,其中每个dna分子包含与所有其他dna分子的第一、第二和第三组分序列相同长度的第一、第二和第三组分序列。
114.实施方式112的方法,其中所述第一、第二和第三组分序列各自在约15bp和约1200bp之间。
115.实施方式112的方法,其中所述第一、第二和第三组分序列各自在约100bp至约600bp之间。
116.实施方式112-115中任一项的方法,其中所述至少三组dna分子中的任一个的第一、第二和第三组分序列都不是相同的序列。
117.实施方式112的方法,其中所述第一和第二限制位点的序列选自ecori位点、bamhi位点、hindiii位点、noti位点、pvuii位点、smai位点、ecorv位点、kpni位点、psti位点、sali位点、scai位点、spei位点、sphi位点、stui位点和xbai位点的序列。
118.实施方式112的方法,其中所述第一和第二限制位点的序列包含xhoi位点,所述限制性酶包含xhoi。
119.实施方式112-118中任一项的方法,其中所述至少三个dna分子中的每一个的序列进一步包含第一引物序列和与第二引物序列互补的序列,其中所述第一引物序列位于所述第一组分序列的5',其中与第二引物序列互补的序列位于第三组分序列的3'。
120.实施方式119的方法,其中所述第一引物序列包含序列5'-tgaagaactgcggcagg-3'(seqidno:12)。
121.实施方式119或120的方法,其中与第二引物序列互补的序列包含序列5'-ggtctgcacttccagct-3'(seqidno:13)。
122.实施方式112-121中任一项的方法,其中所述至少三组消化的dna分子的预定量是相等的。
123.实施方式112-122中任一项的方法,其中所述至少三组消化的dna分子的预定量不相等。
124.实施方式123的方法,其中所述至少三组消化的dna分子的预定量为线性序列或几何序列。
125.实施方式123的方法,其中一组消化的dna分子的预定量是基线,并且所述至少两组另外的消化的dna分子的预定量是基线的整数倍。
126.实施方式123的方法,其中一组dna分子的预定量是基线,并且其中每个其他组的消化的dna分子的预定量是另一组消化的dna分子的预定量的两倍,不包括基线。
127.实施方式123的方法,其中所述至少三组消化的dna分子的预定量的比率为1:2:4。
128.实施方式112-127中任一项的方法,其中(d)的混合步骤还包括将预定量的至少另一组dna分子与来自步骤(a)-(c)的至少三组消化的dna分子混合以在单个归一化对照中产生至少四组dna分子,并且其中至少另一组dna分子从样本中分离或衍生。
129.实施方式128的方法,其中所述样本包含质粒dna,线粒体dna,叶绿体dna或基因组dna。
130.实施方式128或129的方法,其中所述至少四组dna分子的预定量是相等的。
131.实施方式128或129的方法,其中所述至少四组dna分子的预定量不相等。
132.实施方式131的方法,其中所述至少四组dna分子的预定量为线性序列或几何序列。
133.实施方式131的方法,其中一组dna分子的预定量是基线,并且所述其他组的dna分子的预定量是基线的整数倍。
134.实施方式131的方法,其中一组消化的dna分子的预定量是基线,并且其中每个其他组的消化的dna分子的预定量是另一组消化的dna分子的预定量的两倍,不包括基线。
135.实施方式131的方法,其中所述至少四组消化的dna分子的预定量的比率为1:2:4:8。
136.一种制备包含rna分子的归一化对照的方法,所述方法包括:
a.合成至少三组dna分子,其中所述至少三个dna分子中的每一个dna分子从5'至3'包含第一组分序列、与启动子序列互补的序列、第一限制性酶切位点、第二组分序列、与启动子序列互补的第二序列、第二限制性位点、第三组分序列和与启动子序列互补的第三序列;
b.用切割第一和第二限制位点的限制性酶消化所述多组dna分子中的每个组,以产生至少三组经消化的dna分子;
c.体外转录至少三组消化的dna分子中的每一组,以产生至少三组rna分子;
d.对rna分子群体中的每个群体中产生的rna的量进行定量;以及
e.混合预定量的所述rna分子群体中的各群体,从而产生包含rna分子的归一化对照。
137.实施方式136的方法,其中所述至少三个dna分子中的每一个包含与所述至少三个dna分子中的任何其他分子的第一、第二和第三组分序列具有相同长度的第一、第二和第三组分序列。
138.实施方式136的方法,其中第一启动子序列,第二启动子序列和第三启动子序列包含相同的启动子序列。
139.实施方式138的方法,其中所述启动子序列包含选自t7启动子序列,sp6启动子序列或t3启动子序列的启动子序列。
140.实施方式139的方法,其中所述t7启动子序列包含序列5'-taatacgactcactataggg-3'(seqidno:15)。
141.实施方式136-140中任一项的方法,其中所述第一和第二限制位点的序列相同。
142.实施方式136-141中任一项的方法,其中第一、第二和第三组分序列均介于约15bp与约1200bp之间。
143.实施方式136-141中任一项的方法,其中第一、第二和第三组分序列均介于约100bp与约600bp之间。
144.实施方式136-141中任一项的方法,其中所述至少三组dna分子中的任何一个的第一、第二和第三组分序列都不是相同的序列。
145.实施方式136-144中任一项的方法,其中所述第一和第二限制位点的序列选自ecori位点,bamhi位点,hindiii位点,noti位点,pvuii位点,smai位点,ecorv位点,kpni位点,psti位点,sali位点,scai位点,spei位点,sphi位点,stui位点和xbai位点的序列。
146.实施方式145的方法,其中所述第一和第二限制位点的序列包含xhoi位点,并且所述限制性酶包含xhoi。
147.实施方式136-146中任一项的方法,其中所述至少三个rna分子群体中的每一个的预定量是相等的。
148.实施方式136-146中任一项的方法,其中所述至少三个rna分子群体中的每一个的预定量不相等。
149.实施方式148的方法,其中所述至少三个rna分子群体的预定量为线性序列或几何序列。
150.实施方式148的方法,其中一个合成rna分子群体的预定量是基线,并且其中至少两个另外的rna分子群体的预定量是基线的整数倍。
151.实施方式148的方法,其中一个rna分子群体的预定量是基线,并且其中每个其他rna分子群体的预定量是除基线之外的另一个rna分子群体的浓度的两倍。
152.实施方式148的方法,其中所述至少三个rna分子群体的预定量为1:2:4的比率。
153.实施方式136-152中任一项的方法,所述方法进一步包括将归一化对照稀释至约0.005至约500pg/μl之间的浓度。
154.实施方式136-152中任一项的方法,所述方法进一步包括将归一化对照稀释至约0.5至约50pg/μl之间的浓度。
155.实施方式136-152的方法,其中所述归一化对照浓度为1.4pg/μl。
156.一种制备归一化对照的方法,所述归一化对照包括至少三组多核苷酸,其中每组中的多核苷酸具有相同的长度,该方法包括:
i.从样本中提取dna;
ii.用限制性酶消化dna以产生dna片段集合;
iii.分离dna片段集合;
iv.纯化dna片段以产生至少三组多核苷酸,其中每组内的多核苷酸具有相同的长度;和
v.将预定量的所述多组经消化的dna分子中的各个组混合,从而产生归一化对照。
157.实施方式156的方法,其中所述限制性酶选自ecori,bamhi,hindiii,pvuii,smai,ecorv,kpni,psti,sali,scai,spei,sphi,stui,xbai,noti,asci,fsei,paci,pmei,bglii,bstbi,hincii和sgfi。
158.实施方式156或157的方法,其中分离dna片段集合包括基于片段大小的分离。
159.实施方式158的方法,其中基于大小分离dna片段包括凝胶电泳,色谱法或切向流过滤(tff)。
160.实施方式156-159中任一项的方法,所述方法还包括将衔接子连接到所述至少三个不同组的多核苷酸中的每一个。
161.通过实施方式112-160中任一项的方法生成的归一化对照。
162.试剂盒,其包含实施方式1-111中任一项的归一化对照。
163.一种多分析物对照,其包含至少三种不同物种的生物的混合物,其中所述生物已被灭活。
164.实施方式163的多分析物对照,其中每种物种以相同的滴度存在于所述多分析物对照中。
165.实施方式163的多分析物对照,其中每种物种以不同的滴度存在于多分析物对照中。
166.实施方式163的多分析物对照,其中所述滴度为线性序列或几何序列。
167.实施方式163的多分析物对照,其中至少两种物种以相同的滴度存在于所述多分析物对照中,并且至少两种物种以不同的滴度存在于所述混合物中。
168.实施方式163-167中任一项的多分析物对照,其中所述多分析物对照中的每个不同物种以约5至约1000万单位/ml(u/ml)的滴度存在。
169.实施方式168的多分析物对照,其中生物的物种包括病毒,细菌,真菌或真核寄生虫的物种。
170.实施方式169的多分析物对照,其中所述病毒是dna病毒。
171.实施方式169的多分析物对照,其中所述病毒是rna病毒。
172.实施方式169的多分析物对照,其中所述细菌是革兰氏阳性细菌或革兰氏阴性细菌。
173.实施方式163-172中任一项的多分析物对照,其中生物的物种是人类病原体。
174.实施方式173的多分析物对照,其中所述多分析物对照中的每个不同物种的滴度为每ml约5至约1000万感染单位(iu/ml)。
175.实施方式163的多分析物对照,其中所述多分析物对照包含10种或11种病毒或基本上由其组成。
176.实施方式175的多分析物对照,其中所述多分析物对照中的物种选自表8中所列的物种的集合。
177.实施方式173或175的多分析物对照,其中所述人类病原体包括在血液或组织移植物中发现的人类病原体。
178.实施方式177的多分析物对照,其中在血液或组织移植物中发现的人类病原体包括巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人类细小病毒b19(b19),人类免疫缺陷病毒(hiv),乙型肝炎,丙型肝炎,i型和ii型人类t淋巴细胞性病毒(htlv),西尼罗河病毒,寨卡病毒,链球菌种,金黄色葡萄球菌,粪肠球菌,蜡状芽孢杆菌,痤疮丙酸杆菌,粘质沙雷氏菌,沙雷氏菌粘菌,小肠结肠炎耶尔森菌,肠杆菌属,不动杆菌属,假单胞菌属,大肠杆菌,肺炎克雷伯菌,奇异变形杆菌,鲍氏不动杆菌,蜡状芽孢杆菌,凝固阴性葡萄球菌,链球菌,克雷伯菌属,沙雷氏菌属雷氏普罗威登斯菌,苍白密螺旋体,克鲁斯锥虫和小巴贝虫中的一种或多种。
179.实施方式173或174的多分析物对照,其中所述多分析物对照中的人类病原体选自表2-3或5-7。
180.实施方式163-172中任一项的多分析物对照,其中所述物种不是人类病原体。
181.实施方式180的多分析物对照,其中所述物种与作为人类病原体的物种在相同属中。
182.实施方式163-172中任一项的多分析物对照,其中所述物种包括作为人类病原体的物种和不是人类病原体的物种的混合物。
183.实施方式163-182中任一项的多分析物对照,其中所述生物已通过化学处理,热处理,ph处理或紫外线照射而灭活。
184.实施方式183的多分析物对照,其中所述化学处理包括甲醛。
185.实施方式163-184中任一项的多分析物对照,其还包含血浆。
186.实施方式183的多分析物对照,其中所述血浆是人血浆。
187.包含从实施方式163-186中任一项的多分析物对照中提取的核酸的集合。
188.实施方式163-186中任一项的多分析物对照或实施方式185的集合,还包括实施方式1-111中任一项的归一化对照。
189.一种试剂盒,所述试剂盒包含实施方式163-186中任一项的多分析物对照或实施方式187的集合。
190.实施方式189的试剂盒,所述试剂盒进一步包含实施方式1-111中任一项的归一化对照。
191.一种量化样本中的至少一个靶序列表达水平的方法,所述方法包括:
a.将已知量的实施方式1-111中任一项的归一化对照与样本混合,
b.制备高通量测序文库,
c.对所述文库进行测序以产生读段的集合,
d.将来自读段集合的读段映射到样本或归一化对照,
e.确定通过归一化对照中的多核苷酸的每个组或亚组产生的读段的数目,
f.计算与(a)中的样本混合的归一化对照中的多核苷酸的每个组或亚组的初始浓度与(c)中产生的读段的数目之间的关系,
g.模拟样本中的至少一个靶序列的读段与浓度之间的关系,和
h.使用(g)中的模型,由通过靶序列产生的读段的数目计算样本中的至少一种靶多核苷酸的初始浓度。
192.实施方式191的方法,所述方法进一步包括从样本中提取核酸。
193.实施方式191的方法,其中所述核酸包含dna,rna或其混合物。
194.实施方式191-193中任一项的方法,其中(a)的混合步骤发生在(b)的文库制备步骤之前,与(b)的文库制备步骤同时或在(b)的文库制备步骤之后。
195.实施方式191-194中任一项的方法,所述方法进一步包括从样本中提取核酸,并且其中(a)的混合步骤在从样本中提取核酸之前进行。
196.实施方式191-195中任一项的方法,其中将归一化对照中的至少三组多核苷酸中的每组以约0.001:0.420:1的比率加到样本中。
197.实施方式191-196中任一项的方法,其中将归一化对照中的至少三组多核苷酸中的每组以约0.001pg/μl至约5pg/μl之间的最终浓度加到样本。
198.实施方式191-197中任一项的方法,其中所述样本是人样本。
199.实施方式191-198中任一项的方法,其中所述靶序列包含病毒,细菌,真菌,真核寄生虫或一个或多个人类基因的序列。
200.实施方式191-198中任一项的方法,其中所述靶序列包含毒力因子序列,抗生素抗性基因或与致病性相关的序列。
201.实施方式191-198中任一项的方法,其中所述样本包括来自诊断为患有癌症的受试者的样本。
202.实施方式201的方法,其中所述癌症包括细胞群体,所述细胞群体包含相对于所述受试者的非癌细胞具有至少一种遗传变化的序列。
203.实施方式202的方法,其中所述至少一个遗传改变包括单核苷酸多态性(snp),插入,缺失,倒位或重排。
204.实施方式201-203中任一项的方法,其中所述靶序列包含具有至少一个遗传改变的序列。
205.实施方式191-197中任一项的方法,其中所述样本包括宏基因组样本。
206.实施方式205的方法,其中所述宏基因组样本是环境样本。
207.实施方式205或206的方法,其中所述靶多核苷酸包含在宏基因组学样本内的物种的序列。
208.实施方式205-207中任一项的方法,其中所述靶多核苷酸包含毒力因子序列,抗生素抗性基因或与致病性相关的序列。
209.实施方式191-193中任一项的方法,其中所述样本包括多分析物对照,所述多分析物对照包括至少三种不同物种的生物的混合物,其中所述生物已被灭活。
210.实施方式209的方法,其中所述至少三种不同物种的生物中的每一种均包含至少一种靶序列。
211.实施方式210的方法,其中所述至少一种靶序列是每种生物物种中的不同靶序列。
212.实施方式209的方法,其中
(i)在步骤(d)中将来自读段集合的读段映射到样本还包括将读段映射到多分析物对照(d)中的至少三种生物的每一种;和
(ii)在(h)计算至少一个靶序列的初始浓度包括为每种物种计算至少一个靶序列的初始浓度。
213.实施方式209-212中任一项的方法,其中每种物种以相同的滴度存在于多分析物对照中。
214.实施方式209-212中任一项的方法,其中每种物种以不同的滴度存在于多分析物对照中。
215.实施方式214的方法,其中所述滴度为线性序列或几何序列。
216.实施方式209-212中任一项的方法,其中至少两种物种以相同的滴度存在于多分析物对照中,并且至少两种物种以不同的滴度存在。
217.实施方式209-216中任一项的方法,其中所述多分析物对照中的每种物种的滴度为5至1000万单位/ml(u/ml)。
218.实施方式209-217中任一项的方法,其中所述生物是病毒,细菌,真菌,真核寄生虫或其组合。
219.实施方式218的方法,其中所述病毒是dna病毒。
220.实施方式218的方法,其中所述病毒是rna病毒。
221.实施方式218的方法,其中所述细菌是革兰氏阳性细菌或革兰氏阴性细菌。
222.实施方式209-221中任一项的方法,其中所述生物的物种是人类病原体。
223.实施方式222的方法,其中所述多分析物对照中的每个不同物种以约5至约1000万感染单位/ml(iu/ml)的滴度存在。
224.实施方式222或223的方法,其中所述多分析物对照包括10种或11种病毒物种或基本上由其组成。
225.实施方式224的方法,其中所述多分析物对照中的物种选自表8中列出的物种的集合。
226.实施方式222或223的方法,其中所述人类病原体包括在血液或组织移植物中发现的人类病原体。
227.实施方式226的方法,其中在血液或组织移植物中发现的人类病原体包括巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人类细小病毒b19(b19),人类免疫缺陷病毒(hiv),乙型肝炎,丙型肝炎,i型和ii型人类t淋巴细胞性病毒(htlv),西尼罗河病毒,寨卡病毒,链球菌种,金黄色葡萄球菌,粪肠球菌,蜡状芽孢杆菌,痤疮丙酸杆菌,粘质沙雷氏菌,沙雷氏菌粘菌,小肠结肠炎耶尔森菌,肠杆菌属,不动杆菌属,假单胞菌属,大肠杆菌,肺炎克雷伯菌,奇异变形杆菌,鲍氏不动杆菌,蜡状芽孢杆菌,凝固阴性葡萄球菌,链球菌,克雷伯菌属,沙雷氏菌属雷氏普罗威登斯菌,苍白密螺旋体,克鲁斯锥虫和小巴贝虫中的一种或多种。
228.实施方式222或223的方法,其中多分析物对照中的人类病原体选自表2-3或5-7。
229.实施方式209-224中任一项的方法,其中所述生物已通过化学处理,热处理,ph处理或紫外线照射而灭活。
230.实施方式229的方法,其中化学处理包括甲醛。
231.实施方式209-230中任一项的方法,其中所述多分析物对照还包括血浆。
232.实施方式231的方法,其中所述血浆是人血浆。
233.实施方式191-232中任一项的方法,其中所述方法包括将已知量的多分析物对照与样本混合,其中所述多分析物对照包括至少三种不同物种的生物的混合物,并且其中生物已被灭活。
234.实施方式233的方法,其中所述生物是病毒,细菌,真菌,真核寄生虫或其组合。
235.实施方式234的方法,其中所述病毒是dna病毒。
236.实施方式234的方法,其中所述病毒是rna病毒。
237.实施方式234的方法,其中所述细菌是革兰氏阳性细菌或革兰氏阴性细菌。
238.实施方式234-237中任一项的方法,其中所述生物不是人类病原体。
239.实施方式238的方法,其中所述生物是与人类病原体的物种在相同属中的物种。
240.实施方式239的方法,其中所述人类病原体包括在血液或组织移植物中发现的人类病原体。
241.实施方式240的方法,其中在血液或组织移植物中发现的人类病原体包括巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人类细小病毒b19(b19),人类免疫缺陷病毒(hiv),乙型肝炎,丙型肝炎,i型和ii型人类t淋巴细胞性病毒(htlv),西尼罗河病毒,寨卡病毒,链球菌种,金黄色葡萄球菌,粪肠球菌,蜡状芽孢杆菌,痤疮丙酸杆菌,粘质沙雷氏菌,沙雷氏菌粘菌,小肠结肠炎耶尔森菌,肠杆菌属,不动杆菌属,假单胞菌属,大肠杆菌,肺炎克雷伯菌,奇异变形杆菌,鲍氏不动杆菌,蜡状芽孢杆菌,凝固阴性葡萄球菌,链球菌,克雷伯菌属,沙雷氏菌属雷氏普罗威登斯菌,苍白密螺旋体,克鲁斯锥虫和小巴贝虫中的一种或多种。
242.实施方式234-240中任一项的方法,其中所述生物已通过化学处理,热处理,ph处理或紫外线照射灭活。
243.实施方式234-242中任一项的方法,其中将已知量的多分析物对照与样本混合在步骤(b)中的文库制备之前进行。
244.实施方式234-243中任一项的方法,其中将所述多分析物对照以所述多分析物对照中的每种物种的约5至1000万u/ml之间的浓度加到所述样本。
245.实施方式191-244中任一项的方法,其中所述模型是线性模型。
246.实施方式191-244中的任一项的方法,其中所述模型用机器学习分类器创建。
247.实施方式246的方法,其中所述机器学习分类器是受监督的。
248.实施方式247的方法,其中所述机器学习分类器是矢量支持机器。
249.实施方式246的方法,其中样本中的至少一种靶核酸分子的初始浓度与通过靶核酸分子产生的读段的数目的线性回归具有在归一化后大于0.95,大于0.96,大于0.97,大于0.98或大于0.99的r2值。
250.实施方式249的方法,其中线性回归的r2值在归一化后提高至少0.01、0.03、0.05、0.07、0.09、0.1、0.13、0.150.17、0.19、0.2、0.23、0.25、0.27、0.29、0.3,0.33、0.35、0.37、0.39、0.4、0.43、0.45、0.47、0.49、0.5、0.53、0.55、0.57、0.59、0.6、0.63、0.65、0.67、0.69、0.7或其之间的任何值。
251.一种量化样本中的靶生物滴度的方法,所述方法包括:
a.提供包含靶生物的样本,其中所述靶生物包含至少一个靶序列;
b.提供包含至少三种生物物种的已知滴度的多分析物对照,其中所述生物已被灭活;
c.将已知量的实施方式1-111中任一项的归一化对照与样本和多分析物对照混合;
d.从样本和多分析物对照中制备高通量测序文库;
e.对所述文库测序以产生样本读段集合和多分析物对照读段集合;
f.使用归一化对照将样本读段集合和来自(e)的多分析物对照读段集合归一化;
g.确定多分析物对照中的至少三种生物物种的归一化读段与已知滴度之间的关系;和
h.使用(g)中确定的关系计算样本中靶生物的α。
252.实施方式251的方法,其中所述至少三种生物各自包含至少一种种特异性靶序列。
253.实施方式252的方法,其中至少一种物种特异性靶序列在多分析物对照中的每个物种中是不同的。
254.实施方式251的方法,其中在步骤(f)进行归一化包括:
i.将样本读段集合中的读段映射到样本或归一化对照;
ii.将来自多分析物对照读段集合的读段映射到多分析物对照或归一化对照;
iii.确定通过归一化对照中的多核苷酸的每个组或亚组产生的读段的数目,以收集样本读段和收集多分析物对照读段;
iv.计算归一化对照中的多核苷酸的每个组或亚组的初始浓度与样本和多分析物对照在步骤(e)产生的读段的数目之间的关系;和
v.确定样本和多分析物对照中的读段与浓度之间的关系。
255.实施方式251-254中任一项的方法,其中确定(g)的读段与滴度之间的关系包括:
vi.从多分析物对照中的至少三种物种的每一种计算至少一种物种特异性靶序列的初始浓度;和
vii.计算样本中的至少一个靶序列的初始浓度。
256.实施方式251或255的方法,其中步骤(i)的映射包括将样本读段集合映射到参考基因组。
257.实施方式256的方法,其中参考基因组不包含靶序列。
258.实施方式256或257的方法,其中参考基因组是人基因组。
259.实施方式254-258中任一项的方法,其中步骤(ii)的映射进一步包括针对多分析物对照中的至少三种生物物种中的每一种,对物种特异性参考基因组进行映射。
260.实施方式251-259中任一项的方法,所述方法进一步包括在步骤(b)中以2个浓度,3个浓度,4个浓度,5个浓度,6个浓度,7个浓度,8个浓度,9个浓度或10个浓度提供多分析物对照,然后将步骤(c)-(g)应用于步骤(b)中提供的多分析物对照的每种浓度。
261.实施方式251-260中任一项的方法,其中在步骤(g)的多分析物对照中,归一化读段与至少三种生物物种的已知滴度之间的关系是线性关系。
262.实施方式251-261中任一项的方法,其中每种物种以相同的滴度存在于多分析物对照中。
263.实施方式251-261中任一项的方法,其中每种物种以不同的滴度存在于多分析物对照中。
264.实施方式263的方法,其中所述滴度为线性序列或几何序列。
265.实施方式251-261中任一项的方法,其中至少两种物种以相同的滴度存在于多分析物对照中,并且至少两种物种以不同的滴度存在。
266.实施方式251-265中任一项的方法,其中每种物种以5至1000万单位/ml(u/ml)的浓度存在于所述多分析物对照中。
267.实施方式251-266中任一项的方法,其中所述生物是病毒,细菌,真菌或其组合。
268.实施方式267的方法,其中所述病毒是dna病毒。
269.实施方式267的方法,其中所述病毒是rna病毒。
270.实施方式267的方法,其中所述细菌是革兰氏阳性细菌或革兰氏阴性细菌。
271.实施方式251-270中任一项的方法,其中所述生物是人类病原体。
272.实施方式271的方法,其中所述多分析物对照中的每个不同物种以约5至约1000万感染单位/ml(iu/ml)的滴度存在。
273.实施方式271或272的方法,其中所述多分析物对照包括10种病毒或11种病毒或基本上由其组成。
274.实施方式273的方法,其中所述多分析物对照中的物种选自表8中列出的物种的集合。
275.实施方式271或272的方法,其中所述人类病原体包括在血液或组织移植物中发现的人类病原体。
276.实施方式275的方法,其中在血液或组织移植物中发现的人类病原体包括巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人类细小病毒b19(b19),人类免疫缺陷病毒(hiv),乙型肝炎,丙型肝炎,i型和ii型人类t淋巴细胞性病毒(htlv),西尼罗河病毒,寨卡病毒,链球菌种,金黄色葡萄球菌,粪肠球菌,蜡状芽孢杆菌,痤疮丙酸杆菌,粘质沙雷氏菌,沙雷氏菌粘菌,小肠结肠炎耶尔森菌,肠杆菌属,不动杆菌属,假单胞菌属,大肠杆菌,肺炎克雷伯菌,奇异变形杆菌,鲍氏不动杆菌,蜡状芽孢杆菌,凝固阴性葡萄球菌,链球菌,克雷伯菌属,沙雷氏菌属雷氏普罗威登斯菌,苍白密螺旋体,克鲁斯锥虫和小巴贝虫中的一种或多种。
277.实施方式271或272的方法,其中所述多分析物对照中的人类病原体选自表2-3或5-7。
278.实施方式251-270中任一项的方法,其中所述生物包括人类病原体的物种和不是人类病原体的物种的混合物。
279.实施方式251-278中任一项的方法,其中所述生物已通过化学处理,热处理,ph处理或紫外线照射灭活。
280.实施方式279的方法,其中化学处理包括甲醛。
281.实施方式251-280中任一项的方法,其中所述多分析物对照还包括血浆。
282.实施方式281的方法,其中血浆是人血浆。
283.一种制备多个归一化对照寡核苷酸的方法,所述方法包括:
a.从至少一个参考序列产生多个参考序列片段;
b.根据参考序列片段的数目产生至少一个参数的分布;
c.将分布划分为至少5个箱;
d.从至少5个箱中的至少3个箱中选择至少一个参考序列片段;
e.改组至少3个参考序列以产生改组序列;和
f.合成包含改组序列的寡核苷酸;
从而产生多个归一化对照寡核苷酸。
284.实施方式283的方法,其中所述参数包括以下各项中的至少一项:(1)gc百分比,(2)熵,(3)复杂性,(4)eiip或其组合。
285.实施方式283的方法,其中所述参数包括gc含量百分比。
286.实施方式283-285中任一项的方法,其中每个箱代表分布的1%,2%,5%,10%,15%或20%。
287.实施方式283-285中任一项的方法,其中所述参考序列片段选自gc含量分布百分比的第10,第30,第50,第70,第80和90百分位箱。
288.实施方式283-287中任一项的方法,其中步骤(d)包括从至少3个箱中的每一个中选择至少2个参考,并且其中步骤(e)还包括将来自每个箱的改组的参考序列连接。
289.实施方式283-288中任一项的方法,其中所述至少一个参考序列包含至少2,至少10,至少20,至少50,至少100,至少200,至少300,至少400,至少500,至少600,至少700,至少800,至少900,至少1,000,至少1,200,至少1,300,至少1,400,至少1,500,至少1,600,至少1,700,至少1,800,至少1,900,至少2,000,至少2,200,至少2,400,至少2,600,至少2,800,至少3,000,至少4,000,至少5,000,至少6,000,至少7,000,至少8,000,至少9,000或至少10,000个参考序列。
290.实施方式283-288中任一项的方法,其中所述至少一个参考序列包含约2,000个参考序列。
291.实施方式283-290中任一项的方法,其中所述至少一个参考序列包含基因组序列或cdna序列或其组合。
292.实施方式283-291中任一项的方法,其中所述多个参考序列片段包含片段参考序列的约10-100,约10-80,约20-60,约20-40,约20-30或约20-25个连续核苷酸。
293.实施方式283-292中任一项的方法,其中每个归一化对照寡聚物具有至少以下三个:(1)与来自参考序列分布中对应箱的参考序列片段类似的gc含量百分比,(2)类似的熵,(3)类似的eiip,(4)类似的长度或(5)类似的复杂性。
294.一种用于制备多个归一化对照寡核苷酸的方法,所述方法包括:
a.使用滑动窗口从至少一个参考序列产生多个参考序列片段;
b.根据参考序列片段的数目产生至少一个参数的分布;
c.将分布划分为至少5个箱;
d.从至少5个箱中的至少3个中选择至少两个参考序列片段,其中所述至少两个参考序列片段在参考序列中是不连续的,或来自不同的参考序列;
e.从至少3个箱中的每个箱中连接至少两个参考序列片段;和
f.合成包含连接的参考序列片段的寡核苷酸;
从而产生多个归一化对照寡核苷酸。
295.实施方式294的方法,其中所述参数包括以下各项中的至少一项:(1)gc含量百分比,(2)熵,(3)复杂性,(4)eiip或其组合。
296.实施方式294的方法,其中所述参数包括gc含量百分比。
297.实施方式294-296中任一项的方法,其中每个箱代表分布的1%,2%,5%,10%,15%或20%。
298.实施方式294-296中任一项的方法,其中所述参考序列片段选自gc含量百分比分布的第10,第30,第50,第70,第80和第90个百分位箱。
299.实施方式294-298中任一项的方法,其中所述滑动窗包括1bp,2bp,3bp,4bp或5bp的滑动窗。
300.实施方式294-299中任一项的方法,其中所述参考序列片段是对应的参考序列的约15-60,约20-40,约20-30,约15-32,20-32或约25-35个连续核苷酸。
301.实施方式294-299中任一项的方法,其中所述参考序列片段是对应参考序列的29、30、31、32、33或34个连续核苷酸。
302.实施方式294-301中任一项的方法,其中所述参考序列片段小于测序读段的平均长度的一半。
303.实施方式294-302中任一项的方法,其中所述至少一个参考序列包含至少2,至少10,至少20,至少50,至少100,至少200,至少300,至少400,至少500,至少600,至少700,至少800,至少900,至少1,000,至少1,200,至少1,300,至少1,400,至少1,500,至少1,600,至少1,700,至少1,800,至少1,900,至少2,000,至少2,200,至少2,400,至少2,600,至少2,800,至少3,000,至少4,000,至少5,000,至少6,000,至少7,000,至少8,000,至少9,000或至少10,000个参考序列。
304.实施方式294-302中任一项的方法,其中所述至少一个参考序列包含约2,000个参考序列。
305.实施方式294-304中任一项的方法,其中参考序列包含基因组序列或cdna序列。
306.实施方式294-305中任一项的方法,其中每个归一化对照寡核苷酸具有以下至少三个:(1)与来自参考序列分布中的对应箱的参考序列片段类似的gc含量百分比,(2)类似的熵,(3)类似的eiip,(4)类似的长度或(5)类似的复杂性。
307.通过实施方式283-306中任一项的方法产生的归一化对照。
308.一种试剂盒,所述试剂盒包含实施方式307的归一化对照。
309.一种用于设计多个归一化对照多核苷酸序列的系统,所述系统包括:
a.存储计算机可执行指令的计算机可读存储介质,包括:
i.导入至少一个参考序列的指令;
ii.从至少一个参考序列产生多个参考序列片段的指令;
iii.根据参考序列片段的数目产生至少一个参数的分布的指令;
iv.将分布划分为箱的指令;
v.从箱的至少一个子集中选择多个参考序列片段的指令;
vi.改组多个参考序列片段以产生改组序列的指令;和
从而产生多个归一化多核苷酸序列;和
b.处理器,被配置成执行包括以下步骤的步骤:
i.接收包括至少一个参考序列的一组输入文件;和
ii.执行存储在计算机可读存储介质中的计算机可执行指令。
310.一种用于设计多个归一化对照多核苷酸序列的系统,所述系统包括:
存储计算机可执行指令的计算机可读存储介质,包括:
i.导入至少一个参考序列的指令;
ii.从至少一个参考序列产生多个参考序列片段的指令;
iii.根据参考序列片段的数目产生至少一个参数的分布的指令;
iv.将分布划分为箱的指令;
v.从箱的至少一个子集中的每一个中选择至少两个参考序列片段的指令,其中所述至少两个参考序列片段在参考序列中是不连续的,或是从不同的参考序列中选择的;和
vi.连接来自每个箱的至少两个参考序列片段的指令;
从而产生多个归一化多核苷酸序列;和
b.处理器,被配置成执行包括以下步骤的步骤:
i.接收包括至少一个参考序列的一组输入文件;和
ii.执行存储在计算机可读存储介质中的计算机可执行指令。
311.实施方式309或310的系统,其中所述参数包括以下各项中的至少一项:(1)gc含量百分比,(2)熵,(3)复杂性,(4)eiip或其组合。
实施例
为了可以更有效地理解在此公开的发明,下面提供实施例。应当理解,这些实施例仅用于说明性的目的,并且不应解释为以任何方式限制本发明。
实施例1:rna归一化对照
rna归一化对照可以通过转录随机生成dna序列进行。随机生成序列被合成为gblock,具有用于转录的末端t7启动子序列和xhoi限制性位点以线性化随机dna模板。创建以下所示设计的三个版本,分别称为rna_nc_1,rna_nc_2和rna_nc_3。每个版本都是按照以下设计创建的,但是具有不同的随机生成序列:
5'-tgaagaactgcggcagg(seqidno:12)(mito阵列引物)+175bp随机序列(使用randomdnagenerator设计)+t7rna聚合酶启动子+xhoi位点+250bp随机序列(使用随机dna发生器设计)+t7rna聚合酶启动子+xhoi位点+450bp随机序列(使用随机dna发生器设计)+t7rna聚合酶启动子+(ribo阵列引物)ggtctgcacttccagct-3′(seqidno:13)。
ribo阵列引物的序列包含序列5′-agctggaagtgcagacc-3′(seqidno:20)。
t7启动子的序列包含5′-taatacgactcactataggg-3′的序列(seqidno:15)。
设计的总长度为981bp。具有不同随机序列的三个设计版本按gblocks(基因块)测序,分别称为rna_nc_1,rna_nc_2和rna_nc_3。用xhoi消化rna_nc_1,rna_nc_2,rna_nc_3gblock,以产生三个线性dna片段,并用t7rna聚合酶转录得到的三个片段的混合物,以产生三个rnanc。gblockrna_nc_1产生3个rna片段:rna_nc_1.1,rna_nc_1.2,rna_nc_1.3。gblockrna_nc_2产生3个rna片段:rna_nc_2.1,rna_nc_2.2,rna_nc_2.3。gblockrna_nc_3产生3个rna片段:rna_nc_3.1,rna_nc_3.2,rna_nc_3.3。对应于rna_nc_1.1,rna_nc_2.1和rna_nc_3.1ncrna的dna片段的t7转录产生的ncrna包含与mito阵列引物互补的序列,与175bp随机dna序列互补的序列和在转录期间由t7聚合酶引入的末端ggg序列。对应于ncrnarna_nc_1.2,rna_nc_2.2和rna_nc_3.2的dna片段的t7转录产生的ncrna包含与250bp随机序列互补的序列和在转录期间引入的末端ggg序列。对应于ncrnarna_nc_1.3,rna_nc_2.3和rna_nc_3.3的dna片段的t7转录产生的ncrna包含与450bp随机序列互补的序列和在转录期间引入的末端ggg序列。
用于在样本提取期间添加nc对照的方案在图6中示出。稀释三个nc转录反应(rna归一化对照集1、2和3,也称为nc1,nc2和nc3或rna_nc_1,rna_nc_2和rna_nc_3,每个都包含上述三个转录本)的输出20,000x至0.1ng/μl。然后通过混合25μlrna_nc_1、50μlrna_nc_2和50μlrna_nc_3以1:2:4的比率混合三组nc转录本,最终体积为175μl。该nc混合物总共包含9个不同的转录本,3个包含3个不同的175bp随机序列,3个包含3个不同的250bp随机序列,而3个包含3个不同的450bp随机序列。然后,通过将30μl混合物加到270μlave缓冲液中,将nc混合物再稀释10倍,以产生10pg/μlnc混合物。
使用ez1病毒迷你试剂盒(qiagen)从阴性血浆样本(精确诊断)中提取核酸。将8μl10pg/μlnc混合物和52μlave缓冲液加到ez1试剂盒第3行1.5ml试管中。从第3行试管中取出50μl等分试样,并将其加到400μl样本中,从而在提取期间将70pgnc混合物加到样本中。
分析了在提取期间向阴性血浆样本中添加三种不同量的nc混合物的结果。对于“高”量的nc,每个样本添加38.10pgnc混合物(nc1:5.4pg,nc2:10.8pg,nc3:21.8pg)。对于“中等”量的nc,每个样本添加19.04pgnc混合物(nc1:2.7pg,nc2:5.4pg,nc3:10.8pg)。对于“低”量的nc,9。每个样本添加52pgnc混合物(nc1:1.4pg,nc2:2.7pg,nc3:5.4pg)。“低”数量导致nextseq500下一代测序后的读段为4%。基于重复数据删除的读段,转换为文库的“低”nc1的质量为1.076pg,占输入的76%。
图7总结了从精确诊断阴性血浆制备的rna-seq文库中rna归一化对照的表示,其中在核酸提取期间添加nc。每个文库的平均读段输出为23,000,000次读段,人类平均代表率为80%。rna归一化对照(38.10pg)的“高”水平添加使读段平均代表这些文库中的总读段的16%。rna归一化对照(19.04pg)的“中等”水平添加使读段平均代表这些文库中的总读段的6%。rna归一化对照的“低”水平添加(9.52pg)使读段平均代表这些文库中的总读段的4%。在“低”输入下,rna_nc1片段的转化率为76%(1.067pg)。
实施方式2:dna归一化对照
可以使用随机生成序列制备dnanc。下面是dnanc设计的示例。使用不同的随机生成序列制作该设计的三个版本:
5'-tgaagaactgcggcagg(seqidno:12)(mito阵列引物)+使用随机dna产生器的随机序列(200bp)+xhoi位点+使用随机dna产生器的随机序列(300bp)+xhoi位点+使用随机dna产生器的随机序列(500bp)+t7rna聚合酶启动子+(ribo阵列引物)ggtctgcacttccagct-3'(seqidno:13)。
ribo阵列引物的从5'至3'的序列是5'-agctggaagtgcagacc-3'(seqidno:20)。设计的总长度为1048bp。设计的三个版本按gblocks(基因块)的顺序订购,分别称为gblockdna_nc_1,gblockdna_nc_2和gblockdna_nc_3。gblockdna_nc_1的xhoi消化产生3个片段:dna_nc_1.1,dna_nc_1.2和dna_nc_1.3。gblockdna_nc_2的消化产生3个片段:dna_nc_2.1,dna_nc_2.2和dna_nc_2.3。gblockdna_nc_3的消化产生3个片段:dna_nc_3.1,dna_nc_3.2和dna_nc_3.3。片段dna_nc_1.1、dna_nc_2.1和dna_nc_3.1分别包含mito阵列引物的dna序列,200bp的随机序列(各自不同)和xhoi半位点。片段dna_nc_1.2、dna_nc_2.2和dna_nc_3.2包含300bp的随机序列,侧翼有两个xhoi半位点。片段dna_nc_1.3、dna_nc_2.3和dna_nc_3.3包含xhoi半位点,500bp随机序列,t7启动子序列和ribo阵列引物序列。
在提取基因组dna之前,将归一化对照加到样本中。然后将归一化对照与测序文库的其余部分并行索引。
具有归一化对照的dna文库使用i7和i5衔接子的双重索引策略制作。如下所述的具有i7和i5序列的引物以200μm的浓度重新悬浮。5μl的i5和i7引物在neb缓冲器2中以20μl的总体积混合(图8)。然后将i5和i7引物混合物放入热循环仪中,升温至95℃持续1分钟,然后缓慢冷却至20℃。然后将i5/i7混合物在1200μl的总体积中稀释至15μm的浓度(用于1个反应)。将i7/i5衔接子进一步按1:20稀释,以达到750nm的总浓度。
双索引引物
i701-um16引物:5’phos/gatcggaagagcacacgtctgaactccagtcactacaggtcnnnnnnatctcgt(seqidno:21)
i702-um16引物:5’phos/gatcggaagagcacacgtctgaactccagtcacagttacatnnnnnnatctcgt(seqidno:22)
i501-um16引物:aatgatacggcgaccgagatctacacnnnnnnccagtacaacactctttcccta(seqidno:23)
i502-um16引物:5’aatgatacggcgaccaccgagatgtaccannnnnngtgcttatacactctttggctan(seqidno:24)
图9显示在示例性dna文库中用包括的nc测序的nc读段百分比。将nc对照以图12所示的浓度加到样本。
实施例3:量化样本中的非宿主的水平
在该实施例中,确定非宿主与nc的比率用于减去混合的宿主和非宿主核酸样本中非宿主量化的不同宿主背景量的作用。
nc由一组核酸组成,分别是dna或rna或dna和rna二者的混合物。混合各种大小的nc以提供不同长度和基础成分的线性分布,以模拟ngs文库内的不同可变性。nc中包含的这些核酸可以合成制造,或由天然核酸的培养混合物制造。片段可以代表非常特定的大小,核酸组成,或涵盖各种浓度的非常大范围的片段大小。可以包含序列基序以了解文库处理是否因序列重复或重复序列而有偏差
nc是一种或多种生物。例如,nc包含从一种或多种生物或生物物种,例如病毒,细菌,真菌或真核生物中提取的多核苷酸,其模拟混合的宿主/非宿主样本中非宿主序列的一种或多种特性。
nc用于确定样本中的不同核酸的相对量。例如,归一化对照用于确定来自被感染宿主(例如,被病毒或细菌感染的人类宿主)的样本中病原体(非宿主)的丰度值。通过将映射到病原体的ngs读段除以映射到nc的ngs读段的数目来确定病原体的丰度值,以补偿背景宿主的不同含量。该值被映射到标准曲线,该曲线以相同的方式生成和归一化,并从标准曲线推断出病原体滴度。使用这样的方法,不一定需要确定绝对病原体负荷。
nc用于确定样本中的不同核酸的绝对量。例如,nc用于确定来自被感染宿主的样本(例如,来自被病毒或细菌感染的人类宿主的样本)中病原体(非宿主)的绝对量。通过将映射到病原体的ngs读段的数目除以映射到nc的ngs读段的数目来补偿不同的背景宿主含量,可以确定病原体的丰度值。基于该比率和nc的输入量(例如,噬菌体或多核苷酸的拷贝),确定相对病原体基因组拷贝。然后针对病原体基因组大小校正该值以在起始材料中得出“绝对”病原体丰度。
在一些情况下,nc包含t4噬菌体核酸序列,而病原体包含巨细胞病毒。由于t4噬菌体和cmv基因组都在200kb左右,两者的大小大致相同,并且假定噬菌斑形成单位/ml(pfu/ml,t4)和拷贝/ml(cp/ml,cmv)为相等的。如果在下一代测序后,每1份t4噬菌体nc读段存在2份cmv读段,并且以100pfu/ml的浓度加入nc,则cmv的浓度必须为200cp/ml。如果cmv是t4基因组大小的1/2,则cmv和t4在初始样本中的浓度大致相同(100pfu/ml或cp/ml)。
实施例4:使用多分析物对照确定病毒滴度
本公开的归一化对照可以用于确定多分析物对照中的一组病毒滴度,用于计算实验样本中的病毒滴度。
在该实施例中,多分析物对照是人类移植样本中常见的11种病毒的混合物:巨细胞病毒(cmv),爱泼斯坦-巴尔病毒(ebv),腺病毒(adv),bk病毒(bkv),jc多瘤病毒(jcv),人疱疹病毒6a(hhv6a),人疱疹病毒6b(hhv6b),单纯疱疹病毒1型(hsv1),单纯疱疹病毒2型(hsv2),水痘带状疱疹病毒(vzv)和人细小病毒b19(b19)。使用11种病毒的混合物来构建不同滴度(例如0、100、1,000和10,000感染单位(iu)/ml)的线性面板,并掺入血浆中。可以将多分析物对照掺入不同水平的血浆中,以生成多分析物对照的校准面板。将归一化对照加到校准面板中的样本和多分析物对照,提取核酸,然后从实验样本和多分析物对照中生成一个或多个高通量测序文库。在多分析物对照面板或归一化对照中,将读段映射到不同的病毒,并且归一化对照用于对来自多分析物对照中的每种病毒的读段计数进行归一化。然后针对多分析物对照将归一化读段计数相对于滴度绘图以产生标准曲线(图13),并计算归一化读段与log(iu/ml)之间的关系方程。在这个实施例中(图13),这种关系是线性的。将来自实验样本的病毒信号类似地用归一化对照进行归一化,并且从多分析物对照计算的关系用于计算实验样本中的病毒滴度。
该多分析物对照也用作阳性外部对照,它与实验样本分开进行整个样本制备过程。高和低浓度的多分析物对照也可以与实验样本一起包含以确保先前的校准面板(例如参考面板)仍适用于当前条件。
实施例5:使用序列改组生成归一化对照序列
设计归一化对照的一种方法是通过基础改组方法(cantor改组)。该设计过程的图在图14a中显示。在改组方法中,靶序列的所有相同碱基均以相同比率保留,并且靶序列的参数被保留,但归一化对照的序列被改组且未映射到靶序列。
代表潜在病原体的约1200-2000个参考基因组用于生成gc含量、熵、复杂性和电子-离子相互作用潜能(eiip)相对于基因组长度的分布。在图15中显示三元图,其显示参考基因组的gc含量、熵和eiip之间的相互作用。
序列从长度箱获得,长度箱相对于不同分布(gc含量,熵,复杂性,eiip)在x轴(长度)的第10,30,50,70,80和90百分位。每个序列在其内部通过20-25个mer窗口改组,并放置在序列库中以形成潜在阵列。每个序列还通过100个mer窗口在其内部改组,并放置在序列库中,以用作“映射对照”。
将潜在阵列和“映射对照”与参考基因组进行分区,并针对ncbi非冗余序列数据库(nr)和引导序列进行blast比对。消除潜在阵列中映射的序列。归一化对照设计为55个交错浓度。
归一化对照通过信息学方法掺入probit(1000cp/ml),临床样本,识别和量化寡核苷酸,识别和量化病原体。将“映射对照”序列与参考进行映射以证明改组有效。
在图14b-14c中显示以每对数浓度1-3个寡核苷酸设计的归一化对照的示例。在图14d中显示示例性归一化对照,其设计为浓度的每个数量级由6个浓度表示,每个对数浓度具有至少一个不同的寡核苷酸,并且覆盖7-8个数量级。
图16-17显示使用参考基因组面板生成的kmer中eiip、熵和gc含量的分布。生成32亿个kmer,并使用20亿个kmer(图16)、1000万个kmer(图17a)和130万个kmer(图17b)构建初始配置文件。将kmer改组以模拟参考基因组中的eiip分布、熵和gc含量。
python中的多重处理可用于对kmer数据集(例如process和pool)执行处理。pandas和process用于对大文件执行类似的操作。python中的datamash用于12亿行kmer(分为7个文件)以获取功能的百分位值。awk用于执行选择并将选择的kmer写入文件,该文件用于构建寡核苷酸。
在图18中显示用于处理kmer并产生输出寡核苷酸的示例性管线。对kmer进行索引以显示它们已在归一化对照寡核苷酸构建中使用了多少次并启动全局变量dist_range={1kb…10kb}。检查(归一化对照寡核苷酸的)输出数据帧,以查看长度和位置是否在dist_range内,以及生成的寡核苷酸的数量。如果为“否”,则选择未覆盖的长度箱,然后从步骤1中创建的索引中选择使用最少的kmer。将这些连接,直到达到设计的归一化对照长度,然后重复该过程,直到生成所需数量的寡核苷酸。然后将序列传递给shuffle函数。
图19a-19d显示由cantor改组产生的归一化对照模拟bk多瘤病毒分离株ch-1,jc多瘤病毒株niid12-31,人多瘤病毒1株bk2和人腺病毒c的eiip、复杂性和gc含量。图20a-20d显示使用生物技术信息中心(ncbi)blast核苷酸序列服务器(blast.ncbi.nlm.nih.gov/blast.cgi?page_type=blastsearch),归一化对照不会对bk多瘤病毒分离株ch-1,jc多瘤病毒株niid12-31,人多瘤病毒1株bk2和人腺病毒c产生blast。
实施例6:使用滑动窗口生成归一化对照序列(穷举kmer方法)
实施例5中描述的cantor改组的替代方法是使用来自靶序列的自然序列。在这种方法中,在自然序列上采用滑动窗口以生成交错的、重叠的kmer。然后将不相邻的kmer连接以产生归一化对照序列。如果使用约31或32bp的kmer大小,则约150bp的测序读段将包含多个kmer,并且可以通过在靶序列中不相邻的kmer的并列识别归一化对照序列。
在这种方法中,可以通过选择分布在整个基因组中的多个序列来表示单个基因组,该基因组代表该基因组如何片段化。此外,各个归一化对照可以包含来自多种生物的kmer,并匹配多种生物的参数。例如,归一化对照寡核苷酸可以包含长kmer,并且在长kmer内可以是包含来自其他生物的序列的较短kmer。
约1200-2000个参考基因组用于生成交错的kmer,并且如上文实施例5所述计算kmer分布。
首先基于gc含量百分比选择kmer:即,选择一定数量的gc含量百分比以将其包括在归一化对照中。接下来,将处于选择gc含量百分比的kmer连接以在gc箱中生成1千碱基(kb)的寡核苷酸。检查连接序列的gc含量,以及熵。然后将序列通过均聚物过滤器,并进行blast比对(blastn和guideblast)以产生785,000个寡核苷酸,每个寡核苷酸的长度为1kb。
图21a-21c显示在选择的gc含量百分比下产生的kmer的eiip、熵和gc含量的分布。当对参考基因组序列和使用这些方法生成的归一化对照序列进行主成分分析(pca)时,两者在熵、eiip和gc含量之间都显示出类似的相关性(图22)。此外,使用该方法产生的归一化对照的熵、eiip和gc含量与参考基因组序列的熵、eiip和gc含量相关(图23)。另外,进行kolmogorovsmirnov(ks)测试,比较参考基因组序列与归一化对照的熵的概率分布(图24)。
使用ncbiblast核苷酸序列服务器通过blast验证该785,000个归一化对照寡核苷酸序列与参考基因组序列不匹配(图25a-25b)。对于50-80个碱基,归一化对照寡核苷酸的头部总是以约750bp对pseudomonas进行blast处理(图25a)。文件的尾部没有blast命中(图25)。可以对所有785,000个归一化对照寡核苷酸序列进行大规模blast处理,并且只有那些没有blast命中的序列才能被选择用作归一化对照。
序列表
<110>阿克生物公司(arcbio,llc)
e·哈尼斯
v·纳格什
l·g·本利
m·l·卡彭特
<120>用于管理下一代测序中的低样本输入的归一化对照
<130>arcb-006/02wo
<150>62/741,466
<151>2018-10-04
<150>62/832,560
<151>2019-04-11
<160>24
<170>patentin第3.5版
<210>1
<211>22
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>crrna编码序列
<400>1
gttttagagctatgctgttttg22
<210>2
<211>86
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>tracrrna编码序列
<400>2
ggaaccattcaaaacagcatagcaagttaaaataaggctagtccgttatcaacttgaaaa60
agtggcaccgagtcggtgcttttttt86
<210>3
<211>83
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>gna茎环
<400>3
gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagt60
ggcaccgagtcggtgcttttttt83
<210>4
<211>83
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>gna茎环反向互补序列
<400>4
aaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaa60
cttgctatttctagctctaaaac83
<210>5
<211>83
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>gna茎环
<400>5
guuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaagu60
ggcaccgagucggugcuuuuuuu83
<210>6
<211>94
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>gna茎环
<400>6
gttttagagctatgctggaaacagcatagcaagttaaaataaggctagtccgttatcaac60
ttgaaaaagtggcaccgagtcggtgctttttttc94
<210>7
<211>94
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>gna茎环反向互补序列
<400>7
gaaaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttatttta60
acttgctatgctgtttccagcatagctctaaaac94
<210>8
<211>94
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>gna茎环
<400>8
guuuuagagcuaugcuggaaacagcauagcaaguuaaaauaaggcuaguccguuaucaac60
uugaaaaaguggcaccgagucggugcuuuuuuuc94
<210>9
<211>19
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>cpf1蛋白结合序列
<400>9
aauuucuacuguuguagau19
<210>10
<211>19
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>编码cpf1蛋白结合序列
<400>10
aatttctactgttgtagat19
<210>11
<211>86
<212>rna
<213>人工序列(artificialsequence)
<220>
<223>tracrrna序列
<400>11
ggaaccauucaaaacagcauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaa60
aguggcaccgagucggugcuuuuuuu86
<210>12
<211>17
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>引物序列
<400>12
tgaagaactgcggcagg17
<210>13
<211>17
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>引物序列
<400>13
ggtctgcacttccagct17
<210>14
<211>19
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>t7启动子
<400>14
taatacgactcactatagg19
<210>15
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>t7启动子
<400>15
taatacgactcactataggg20
<210>16
<211>29
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>t7启动子
<400>16
gcctcgagctaatacgactcactatagag29
<210>17
<211>18
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sp6启动子
<400>17
atttaggtgacactatag18
<210>18
<211>24
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>sp6启动子
<400>18
catacgatttaggtgacactatag24
<210>19
<211>18
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>t3启动子
<400>19
aattaaccctcactaaag18
<210>20
<211>17
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>引物序列
<400>20
agctggaagtgcagacc17
<210>21
<211>54
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>i701-um16引物
<220>
<221>misc_特征
<222>(42)..(47)
<223>n是a、c、g、或t
<400>21
gatcggaagagcacacgtctgaactccagtcactacaggtcnnnnnnatctcgt54
<210>22
<211>54
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>i702-um16引物
<220>
<221>misc_特征
<222>(42)..(47)
<223>n是a、c、g、或t
<400>22
gatcggaagagcacacgtctgaactccagtcacagttacatnnnnnnatctcgt54
<210>23
<211>54
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>i501-um16引物
<220>
<221>misc_特征
<222>(27)..(32)
<223>n是a、c、g、或t
<400>23
aatgatacggcgaccgagatctacacnnnnnnccagtacaacactctttcccta54
<210>24
<211>58
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>i502-um6引物
<220>
<221>misc_特征
<222>(30)..(35)
<223>n是a、c、g、或t
<220>
<221>misc_特征
<222>(58)..(58)
<223>n是a、c、g、或t
<400>24
aatgatacggcgaccaccgagatgtaccannnnnngtgcttatacactctttggctan58
- 还没有人留言评论。精彩留言会获得点赞!