一种双启动子表达载体及其构建方法与流程
本申请属于基因工程技术领域,具体涉及一种能同时在原核及哺乳动物细胞中分别表达目的基因的双启动子表达载体及其构建方法专利申请。
背景技术:
基因工程技术常规应用中,通过将人为获取的外源目的基因与载体在体外进行重组,然后将重组载体导入到受体细胞并使该目的基因在受体细胞中进行正常的复制、转录和翻译,进而可对目的基因功能进行深入分析和研究。基于这一原理,基因工程技术在医药学领域已经得到广泛应用,如生产重组蛋白药物、基因治疗等。
现有技术中,利用基因工程进行重组蛋白药物生产时,依据受体细胞基因组差异,可将蛋白表达系统分为两类:真核表达系统和原核表达系统,其中真核表达系统如哺乳动物细胞、昆虫细胞、酵母细胞等;原核表达系统如大肠杆菌等。其中哺乳动物和大肠杆菌表达系统是现有重组蛋白药物制备中最为常用和最为主要的表达系统。统计表明,现有80%以上批准上市的重组蛋白药物是在这两个表达系统生产的。
现有技术中,用于表达重组蛋白的哺乳动物细胞主要包括:中国仓鼠卵巢细胞(chinesehamsterovariancells,cho)细胞、小鼠骨髓瘤细胞sp2/0和ns0、人胚胎肾细胞hek293、仓鼠肾细胞bhk-21等。而原核表达系统中的大肠杆菌因其遗传背景清楚,具有相对于cho细胞其表达量较高、能在培养基上快速繁殖、成本低等优点,因此以大肠杆菌构建的蛋白表达系统是最常用的原核表达系统。
由于现有原核、真核两个表达系统在表达蛋白时,针对目的基因进行有效复制、选择、转录以及翻译所需的元件不同,因此若想在这两个系统中表达相同的蛋白往往需要针对性选择或设计不同的重组表达载体。而这个过程由于操作步骤较为繁琐、成本较高。
技术实现要素:
本申请目的在于提供一种同时包含真核启动子cmv和原核启动子t7的双启动子表达载体,从而可以在真核细胞或原核细胞中表达相同目的基因,从而为相关基因研究或目的蛋白制备奠定一定技术基础。
本申请所采取的技术方案详述如下。
一种双启动子表达载体,其基因组中包含有原核启动子t7序列、核糖体结合位点rbs和t7终止序列;具体构建时,以pires-neo、pires-neo2、pires-neo3、pegfp-c1、pcdna1.1、pcho1.0等真核表达载体为出发载体,利用基因工程技术构建获得;
所构建的双启动子表达载体中,t7启动子序列位于真核表达载体的启动子(真核启动子例如为:sv40、cmv、ef1α、cag等)下游,核糖体结合位点位于t7启动子序列下游,t7终止序列位于出发载体的真核表达载体筛选标记(例如新霉素磷酸转移酶(neomycinphosphotransferase,npt)抗性基因)下游;
具体构建时,通过将启动子t7序列、核糖体结合位点(rbs)和t7终止序列或者启动子t7序列与核糖体结合位点(rbs)的连接序列、t7终止序列重组至真核表达载体构建获得;
所述启动子t7序列为:taatacgactcactata,
所述rbs序列为:gaagga;
所述启动子t7序列与核糖体结合位点(rbs)的连接序列为:taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattttgtttaactttaagaaggagatatacc;
所述t7终止序列为(48bp):
ctagcataaccccttggggcctctaaacgggtcttgaggggttttttg。
以pires-neo作为出发载体、以egfp基因或asfvp54基因作为目的基因为例,双启动子表达载体构建方法包括如下步骤:
(1)针对目的基因,设计引物并进行pcr扩增
针对egfp基因,以pegfp-c1质粒中的egfp基因序列作为目的基因为例,pcr扩增时可设计引物p1、p2如下:
p1:5′-ccggaattcatggtgagcaagggcgaggag-3′,
p2:5′-ctaggatcccttgtacagctcgtccatgccga-3′;
针对asfvp54基因,所述asfvp54基因序列(576bp)为:
atggattctgaattttttcaaccggtttatccgcggcattatggtgagtgtttgtcaccagtctctacaccaagcttcttctccacacatatgtatactattctcattgctatcgtggtcttagtcattattatcatcgttctaatttatctattttcttcaagaaagaaaaaagctgctgccgctattgaagaggaagatatacagtttataaatccttatcaagatcagcagtgggtagaggtcactccacaaccaggtacctctaaaccggctggagtgactacagcaagtgtaggcaagccagtcacgggcaggccggcaacaaacagaccagttacggacaggctagtcatggtaactggcgggccagcggccgcaagtgcggccgcaagtgcggctgcgaatgcggccacgaatacagctgcgagtgcagccgcgagtgctcctgctcatccggctgagccttatacgacagtcactactcagaacactgcttcccaaacaatgtcggctattgaaaatctacggcaaagaagcacctatacgcataaagacctagaaaactccttgtaa
(2)构建含有目的基因的重组真核表达质粒
对出发质粒pires-neo进行酶切(例如利用ecori、bamhi进行双酶切),回收线性化质粒dna(即线性化的pires-neo质粒);
利用t4dna酶将步骤(1)目的基因序列与线性化质粒dna进行连接,并转化、筛选获得重组后的含有目的基因的重组质粒;
(3)制备启动子t7序列、核糖体结合位点(rbs)和t7终止序列
人工合成或采用其他生物技术分别获得启动子t7序列、核糖体结合位点(rbs)、t7终止序列,或者启动子t7序列与核糖体结合位点(rbs)的连接序列、t7终止序列,备用;
所述启动子t7序列与rbs的连接序列为:
taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattttgtttaactttaagaaggagatatacc;
(上述序列中的1~17位序列,即“taatacgactcactata”为t7启动子,74~80位序列,即“gaagga”部分序列为rbs序列)
所述t7终止序列为(48bp):
ctagcataaccccttggggcctctaaacgggtcttgaggggttttttg;
(4)连接、转化和筛选,构建获得双表达载体
将步骤(3)中的启动子t7序列、核糖体结合位点(rbs)和t7终止序列重组至步骤(二)的含有目的基因的重组质粒中,筛选、验证确保重组构建正确,即为可以在原核或真核哺乳动物表达系统均能表达目的基因的双启动子表达质粒。
利用所述双启动子表达质粒转染细胞时:转染真核哺乳动物细胞时,具体例如采用:cho、hek293、bhk、sp2/o、ns0等细胞株;转化原核细胞时,具体例如采用大肠杆菌bl21细胞株。
本申请所构建的双启动子表达载体,其中原核表达的t7启动子来源于t7噬菌体,是一种大肠杆菌表达系统常用的强启动子,它能够对t7rna聚合酶有特异性反应,被识别后的转录非常活跃。基于此,发明人将其与真核表达启动子进行融合,从而构建获得了一种能在原核和真核细胞中同时表达同一目的基因的双启动子表达载体,从而可以克服现有常规的同一目的蛋白在哺乳动物及原核细胞需要2套表达系统的繁杂步骤,同时也为其它目的基因在不同表达系统中表达奠定一定技术基础。
附图说明
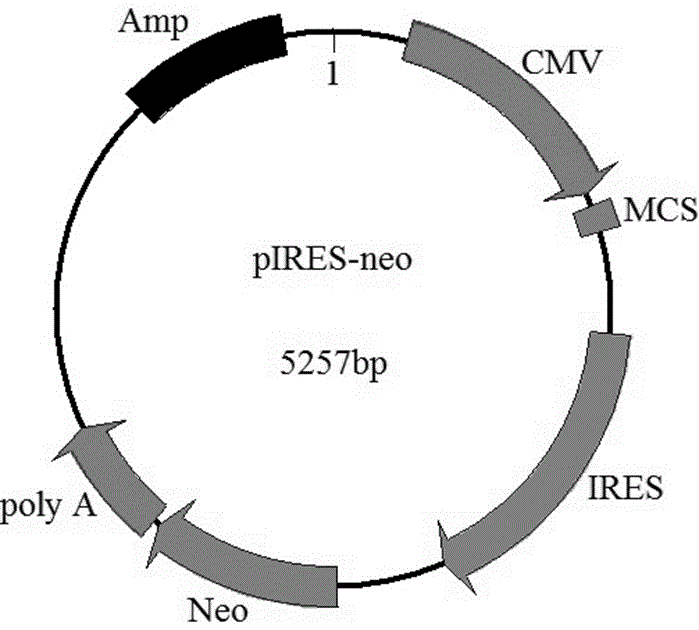
图1为pires-neo载体的结构示意图;
图2为pires-egfp载体的结构示意图;
图3为pires-cmv/t7-egfp载体的结构示意图;
图4为egfp在cho细胞(左图)及原核大肠杆菌bl21中表达荧光图(右图);
图5为流式细胞术检测egfp在cho细胞中的表达水平;
图6为westernblot检测egfp在大肠杆菌bl21细胞中的表达水平;
图7为目的蛋白p54在cho细胞(左图)及原核大肠杆菌bl21中表达(右图)的westernblot检测结果。
具体实施方式
下面结合实施例对本申请做进一步的解释说明。在介绍具体实施例前,就下述实施例中部分实验背景情况简要介绍说明如下。
生物材料:
pires-neo载体、pegfp-c1质粒,购自美国clontech公司;
相关引物序列、基因序列合成及测序均由通用生物基因(安徽)有限公司提供完成;
实验试剂:
无缝克隆试剂盒,novoprotein科技有限公司产品。
实施例1
本实施例先以egfp基因作为目的基因、以pires-neo作为出发载体为例,就本申请所述的双表达载体pires-cmv/t7-egfp构建过程介绍如下。
(一)针对目的基因egfp,设计引物并进行pcr扩增
以pegfp-c1质粒中的egfp(enhancedgreenfluorescentprotein)基因(genbank:u55763.1,第613-1329位碱基)序列作为目的基因,设计引物p1、p2如下:
p1:5′-ccggaattcatggtgagcaagggcgaggag-3′,(5′端gaattc部分序列为引入的ecori酶切位点)
p2:5′-ctaggatcccttgtacagctcgtccatgccga-3′;(5′端ggatcc部分序列为引入的bamhi酶切位点)
以pegfp-c1质粒作为模板,进行pcr扩增(扩增产物大小为717bp),电泳检测后,回收pcr扩增产物获得egfp序列。
pcr扩增时,25μl扩增体系设计如下:
10×pcrbuffer,2.5μl;
引物p1、p2,各1.0μl(10μmol/l);
dntp,2.0μl(25μmol/l);
pegfp-c1质粒模板,1.0μl(100ng/μl);
taq酶,0.5μl(5u/μl);
ddh2o,17μl;
pcr扩增程序为:95℃、3min;94℃、40s,58℃、30s,72℃、40s,每个退火温度4个循环,最后55℃,30个循环,72℃、3min。
(二)构建含有egfp的重组真核表达质粒
利用ecori、bamhi酶分别对步骤(1)中pcr扩增产物(即扩增获得的目的基因egfp序列)和出发质粒pires-neo进行双酶切,电泳检测并回收酶切后的egfp序列和线性化质粒dna(即线性化的pires-neo质粒)。
酶切过程中,20μl酶切体系设计如下:
10×mbuffer,2μl;
ecori、bamhi酶,各0.5μl(10u/μl);
pcr扩增产物(即egfp序列)或线性化质粒dna,1.18μl(0.85μg/μl,针对pcr扩增产物)或1.23μl(0.81μg/μl,针对线性化质粒dna);
补足双蒸水至20μl;
充分混匀后,37℃酶切(针对pcr扩增产物,酶切6h;针对线性化质粒dna,酶切3h)。
对于所回收的egfp序列和线性化质粒dna,利用t4dna酶进行连接。
连接时,20μl连接体系设计如下:
2×quickligationbuffer,10μl;
pires-neo线性质粒dna,200ng;
酶切后的egfp序列片段,87.2ng;
t4连接酶,1μl(350u/μl);
补足双蒸水至20μl;
16℃连接过夜。
将连接产物加入e.colijm109感受态细菌悬液中进行转化。转化结束后,取100μl转化菌液接种在含有氨苄青霉素的lb固体培养板上,37℃培养过夜以进行筛选。最后挑取阳性单菌落摇菌继代培养后,提取细菌质粒进行酶切验证和测序验证,将构建正确的重组质粒命名为pires-egfp(图1为pires-neo载体的结构示意图,图2为pires-egfp载体的结构示意图,可以看出,pires-neo载体中为cmv启动子,重组载体中,egfp序列插入了cmv启动子的下游多克隆位点)。
(三)制备启动子t7序列、核糖体结合位点(rbs)和t7终止序列
人工合成启动子t7序列与核糖体结合位点(rbs)的连接序列、t7终止序列,备用;
所述启动子t7序列与rbs的连接序列为(87bp):
taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattttgtttaactttaagaaggagatatacc;
所述t7终止序列为(48bp):
ctagcataaccccttggggcctctaaacgggtcttgaggggttttttg。
(四)构建获得双表达载体
利用无缝克隆试剂盒,将步骤(三)中的t7+rbs序列、t7终止子序列重组到步骤(二)的pires-egfp中;并将所构建重组载体,转化e.colijm109感受态细菌,筛选、并提取质粒进行双酶切(smai/paci)验证和测序验证,将构建正确的质粒重新命名为pires-cmv/t7-egfp,此即为含有双启动子的重组质粒(重组质粒结构示意图如图3所示,可以看出,在cmv启动子下游插入t7启动子和核糖体结合位点,在neo下游插入t7终止序列)。
实施例2
为确定目的基因egfp在真核细胞或原核细胞中的表达情况,针对实施例1所构建的双启动子表达载体pires-cmv/t7-egfp重组质粒,分别进一步将其转染cho-s细胞和转化大肠杆菌bl21,具体实验过程简介如下。
(一)表达载体转染cho细胞表达系统
预先将cho-s细胞培养于含有体积分数为10%的胎牛血清和1%的青/链霉素的dmem-f12培养基中,培养条件为:37℃、5%co2培养箱中培养,待细胞贴壁生长密度达到90%时,0.25%胰酶消化并收集细胞,以2×105/孔接种于24孔板内,待细胞密度达到(大约24h)60%~70%时,以lipo2000(lipofectamine®2000)作为转染试剂进行转染。
转染过程中,以pires-egfp载体(实施例1中的步骤(二)所构建)为对照组。
具体转染操作为:
将2μllipofectamine2000+50μl无血清dmem-f12培养基在37℃孵箱中静置5min;同时将无血清dmem-f12培养基50μl与1μg表达载体pires-cmv/t7-egfp混合均匀,室温静置20min;
同步地,在24孔培养板上用pbs清洗三遍后,细胞中加入500μl无血清的dmem-f12细胞培养基;
随后,将上述静置后的脂质体与pires-cmv/t7-egfp质粒dna的混合液逐滴轻滴入孔中,尽快轻轻摇晃培养平板使其混匀;
再后,置于5%co2细胞培养箱中,37℃培养6h,随后将无血清dmem-f12培养基更换成dmem-f12完全培养基,细胞培养箱内继续培养。
细胞转染48h后,荧光显微镜下观察转染效率及egfp表达情况,结果发现双启动子载体在cho细胞中大量表达(图4左图),且转染效率与pires-egfp载体无明显差别,这一结果表明,目的蛋白在真核表达系统中的转染效率和瞬时表达不受原核启动子的影响。
之后胰蛋白酶消化并收集细胞,转入细胞培养瓶中继续培养,待细胞长满瓶后,胰酶消化收集细胞,流式细胞仪分析egfp表达量。根据其捕获的每个细胞的荧光强度获得平均荧光强度(meanfuorescenceintensity,mfi),检测结果见图5。分析可以看出:双启动子载体在cho细胞表达的平均荧光强度为1.33×104,而pires-egfp载体为1.48×104,二者表达量无明显差别,说明目的蛋白在真核表达系统中的表达不受原核启动子的影响。
(二)表达载体转化大肠杆菌表达系统
将实施例1中所构建的pires-cmv/t7-egfp重组质粒1μg转化50μl大肠杆菌bl21感受态细菌,然后加入500μllb培养基37℃摇床扩大培养1h,随后涂板于含氨苄青霉素的lb固体培养基上,37℃培养箱中过夜培养。
挑取阳性单克隆加入lb培养基在37℃摇床过夜活化后,取60μl菌液于6ml的lb液体培养基,37℃摇床继续培养,期间不断检测od值,当od为0.6-0.8时加入iptg(终浓度为1mmol/l)继续培养6h,收集菌液。转化过程中,同步设置pires-egfp表达载体作为对照。
将所收集的1ml菌液利用紫外分光光度计测定其od值,并12000rpm离心5min弃上清,根据od值加入pbs重悬菌液。最后置于六孔板中在倒置荧光显微镜下观察egfp表达。结果如图4(右图)所示,可以看出,双启动子载体在原核表达系统中有大量目的蛋白的表达。这一结果表明,重组后质粒可以在原核系统中正常启动目的基因的表达。
参考前述操作或者现有技术,对1ml菌液离心并利用pbs重悬后,进行超声破碎细菌,再次12000rpm离心5min,收集上清,同时沉淀加入等量的pbs重悬。分别加入蛋白上样缓冲液100℃煮样10min,进行westernblot分析,结果见图6。可以看出,双启动子载体在原核表达系统中可以大量表达目的蛋白,而对照载体(即无t7启动子的载体)检测不到目的蛋白的表达,也即,单纯的真核启动子在原核表达系统中无法启动目的蛋白表达。
实施例3
在实施例2基础上,为进一步考察本申请所构建双启动子载体是否适用于其他目的蛋白表达,发明人进一步以asfvp54作为目的蛋白,以此为例,构建了重组的双启动子载体pires-cmv/t7-p54,并分别利用真核表达系统和原核表达系统进行了翻译表达,具体过程简介如下。
所述目的蛋白asfvp54对应的基因序列(576bp),具体如下:
atggattctgaattttttcaaccggtttatccgcggcattatggtgagtgtttgtcaccagtctctacaccaagcttcttctccacacatatgtatactattctcattgctatcgtggtcttagtcattattatcatcgttctaatttatctattttcttcaagaaagaaaaaagctgctgccgctattgaagaggaagatatacagtttataaatccttatcaagatcagcagtgggtagaggtcactccacaaccaggtacctctaaaccggctggagtgactacagcaagtgtaggcaagccagtcacgggcaggccggcaacaaacagaccagttacggacaggctagtcatggtaactggcgggccagcggccgcaagtgcggccgcaagtgcggctgcgaatgcggccacgaatacagctgcgagtgcagccgcgagtgctcctgctcatccggctgagccttatacgacagtcactactcagaacactgcttcccaaacaatgtcggctattgaaaatctacggcaaagaagcacctatacgcataaagacctagaaaactccttgtaa
参考前述操作及现有技术,利用无缝克隆试剂盒,将人工合成的asfvp54基因序列替换pires-cmv/t7-egfp载体中的egfp序列,构建获得重组载体,进一步转化e.colijm109感受态细菌,筛选、并提取质粒进行测序验证后,将构建正确的质粒重新命名为pires-cmv/t7-p54。
进一步地,发明人将pires-cmv/t7-p54质粒分别在原核和真核表达系统中进行了表达验证,具体过程简介如下。
(1)在cho细胞中的表达
预先将cho-s细胞培养于含有体积分数为10%的胎牛血清和1%的青/链霉素的dmem-f12培养基中,培养条件为:37℃、5%co2培养箱中培养。6孔板内以3×106/孔细胞量接种细胞,24小时后细胞达到约70~80%融合度时以lipo2000为转染试剂进行转染。
细胞转染48h后,pbs洗涤、胰酶消化、收集细胞,再加入无血清培养基2~3ml并120rpm条件下进行悬浮培养;培养3天后转入125ml悬浮培养瓶中,初始细胞量为5~6×106个/ml,加入30ml无血清培养基,120rpm悬浮培养5天;
培养结束后,收集细胞,加入细胞裂解液裂解细胞;
将80μl裂解液与20μl蛋白上样缓冲液混合均匀后,100℃煮样10min,westernblot检测asfvp54的表达,实验结果见图7(右图)。可以看出,双启动子载体pires-cmv/t7-p54在cho表达系统中成功表达了目的蛋白asfvp54。
(2)在大肠杆菌中的表达
将pires-cmv/t7-p54重组质粒1μg转化50μl大肠杆菌bl21感受态细菌,转化后,挑取阳性单克隆加入lb培养基,在37℃摇床过夜活化后,取50μl菌液于5ml的lb液体培养基中扩大培养,200rpm/min、37℃摇床培养,培养4h之后测od为0.6~0.8时加入iptg,使iptg终浓度为1mmol/l,再继续培养6h以诱导蛋白表达。
培养结束后,收集菌液进行westernblot检测。实验结果见图7。可以看出双启动子载体pires-cmv/t7-p54在大肠杆菌表达系统中亦成功表达了目的蛋白。
综上结果可以看出,本申请所构建的双启动子载体既可以在真核表达系统中有效表达目的蛋白,也可在原核系统中有效表达,而基于此,可为相关蛋白制备奠定良好的技术基础。
序列表
<110>新乡医学院
<120>一种双启动子表达载体及其构建方法
<130>none
<160>3
<170>siposequencelisting1.0
<210>1
<211>87
<212>dna
<213>bacteriophage
<400>1
taatacgactcactataggggaattgtgagcggataacaattcccctctagaaataattt60
tgtttaactttaagaaggagatatacc87
<210>2
<211>48
<212>dna
<213>bacteriophage
<400>2
ctagcataaccccttggggcctctaaacgggtcttgaggggttttttg48
<210>3
<211>576
<212>dna
<213>africanswinefevervirus
<400>3
atggattctgaattttttcaaccggtttatccgcggcattatggtgagtgtttgtcacca60
gtctctacaccaagcttcttctccacacatatgtatactattctcattgctatcgtggtc120
ttagtcattattatcatcgttctaatttatctattttcttcaagaaagaaaaaagctgct180
gccgctattgaagaggaagatatacagtttataaatccttatcaagatcagcagtgggta240
gaggtcactccacaaccaggtacctctaaaccggctggagtgactacagcaagtgtaggc300
aagccagtcacgggcaggccggcaacaaacagaccagttacggacaggctagtcatggta360
actggcgggccagcggccgcaagtgcggccgcaagtgcggctgcgaatgcggccacgaat420
acagctgcgagtgcagccgcgagtgctcctgctcatccggctgagccttatacgacagtc480
actactcagaacactgcttcccaaacaatgtcggctattgaaaatctacggcaaagaagc540
acctatacgcataaagacctagaaaactccttgtaa576
- 还没有人留言评论。精彩留言会获得点赞!