丙肝病毒NS5B聚合酶抑制剂BMT-052关键中间体的高效合成方法与流程
本发明属于有机合成技术领域及药物应用技术领域,尤其是一种丙肝病毒ns5b聚合酶抑制剂bmt-052关键中间体的高效合成方法。
背景技术:
2014年美国bms公司报道了一类新型四取代的7-氮杂苯并呋喃类化合物。研究发现该类化合物是第二代泛基因型丙肝病毒ns5b聚合酶抑制剂,具有很好的生物活性。此后,经过大量的构效关系研究发现了bms-986139为临床前候选药物。进一步的研究确定了bmt-052是一种可能的临床候选药物,它能够克服以前类似物代谢稳定性差的缺点。合成该类化合物的共用及重要中间体i的结构如下所示。以i作为原料,分别经过3步或4步反应生成bms-986139或bmt-052。
从绿色化学角度来看,bms报道的i的合成路线(如下所示)存在着很多问题。首先,合成效率低,6步总产率仅为16%,限制了工业化生产该类药物的可能性。第二,合成中用了较多的高毒性的含有卤素的试剂。六步合成中前五步反应都用到了含有卤素的试剂。这样不仅环境污染严重,而且原子经济性低。第三,合成中两步反应用了造价昂贵的钯催化剂。最后,合成的最后一步需要用到高毒性的co气体,而且需要高压操作。因此,发展一种产率高,环境污染小的i的合成路线成为解决生产该类药物的关键技术。
技术实现要素:
本发明的目的在于克服现有技术的不足之处,提供bmt-052关键中间体i的高产率、低污染的合成方法。该方法工艺简单,反应条件温和,生产成本低,环境污染小。合成中无需过渡金属催化,且不需要任何保护基。
本发明解决其技术问题采用的技术方案是:
一种丙肝病毒ns5b聚合酶抑制剂bmt-052关键中间体i的高产率合成方法,包括以下步骤g:使化合物6生成i;
而且所述化合物6生成i是与甲胺盐酸盐,diea,hbtu反应下生成的。
而且所述方法进一步包括步骤f:使化合物5反应生成化合物6;
而且所述化合物5生成化合物6是与氢氧化钠溶液发生水解反应生成的。
而且所述方法进一步包括步骤e:使化合物4反应生成化合物5;
而且所述化合物4生成化合物5是与双(三甲基硅基)胺基锂,对氟苯甲酰氯,dmap反应生成的。
而且所述方法进一步包括步骤d:使化合物3发生氧化反应反应生成化合物4;
而且所述化合物3生成化合物4是与过氧化脲,三氟乙酸酐反应生成的。
而且所述方法进一步包括步骤c:使化合物2反应生成化合物3;
而且所述化合物2生成化合物3是与二氯亚砜发生反应生成的。
而且所述方法进一步包括步骤b:使化合物1合成化合物2;
而且所述化合物2生成化合物3是与氢氧化锂,tmscn发生反应生成的。
而且所述方法进一步包括步骤a:使2-氯-3溴-5-甲基吡啶反应生成化合物1;
而且所述方法的合成路线如下:
而且所述方法的具体步骤如下:
步骤a:化合物1的合成:
在氩气保护下,在干燥的双口瓶中依次加入2-氯-3溴-5-甲基吡啶,二氯乙烷,n-溴代丁二酰亚胺和过氧化苯甲酰。其中所述2-氯-3溴-5-甲基吡啶∶二氯乙烷∶n-溴代丁二酰亚胺∶过氧化苯甲酰的比例mmol∶ml∶mmol∶mmol为24.2∶70∶36.3∶2.4,反应液在93℃下反应6小时,反应完全后,冷却至室温后用水淬灭,然后用乙酸乙酯萃取浓缩得到粗产物。通过柱层析纯化得到所需产物为白色固体,即为化合物3-bromo-5-(bromomethyl)-2-chloropyridine;
其中柱层析纯化使用石油醚,乙酸乙酯。石油醚∶乙酸乙酯的体积比100∶1~50∶1
步骤b:化合物2的合成:
在氩气保护下,在干燥的双口瓶中依次加入化合物1,无水乙腈中,一水和氢氧化锂。其中所述化合物1∶无水乙腈∶一水和氢氧化锂∶tmscn的比例mmol∶ml∶mmol∶mmol为19.1∶76∶23.0∶23.0,反应液在常温下反应4小时,反应完毕后用乙酸乙酯淬灭,加水萃取。然后用乙酸乙酯萃取水相3次。浓缩得到粗产物。通过柱层析纯化得到所需产物为白色固体,即为化合物2-(5-bromo-6-chloropyridin-3-yl)acetonitrile;
其中柱层析纯化使用石油醚,乙酸乙酯。石油醚∶乙酸乙酯的体积比20∶1~5∶1
步骤c:化合物3的合成:
在氩气保护下,在干燥的双口瓶中加入化合物2,甲醇,降温至0℃,缓慢滴加二氯亚砜。其中所述化合物2∶甲醇∶二氯亚砜的比例mmol∶ml∶mmol为18.2∶15∶45.5,反应液在常温下反应过夜,反应完毕后使用真空旋转蒸发仪蒸干反应液,随后用饱和碳酸氢钠与二氯甲烷萃取。并旋蒸浓缩得到粗产物。通过柱色谱法纯化得到所需产物为白色固体,即为化合物methyl2-(5-bromo-6-chloropyridin-3-yl)acetate;
其中柱层析纯化使用石油醚,乙酸乙酯。石油醚∶乙酸乙酯的体积比30∶1~10∶1
步骤d:化合物4的合成:
在氩气保护下,在干燥的双口瓶中加入化合物3,二氯甲烷,降温至0℃,再加入过氧化脲,三氟乙酸酐,其中所述化合物3∶二氯甲烷∶过氧化脲∶三氟乙酸酐的比例mmol∶ml∶mmol∶mmol为19.4∶78∶80.1∶72.7,反应液在常温下反应1h,反应完毕后使用饱和亚硫酸钠淬灭,二氯甲烷萃取。并旋蒸浓缩得到产物为白色固体,即为化合物3-bromo-2-chloro-5-(2-methoxy-2-oxoethyl)pyridine1-oxide。
步骤e:化合物5的合成:
在氩气保护下,在干燥的双口瓶中加入化合物4,无水四氢呋喃,将反应体系降至-78℃,然后滴加双(三甲基硅基)胺基锂,在-78℃下反应2h,随后缓慢滴加对氟苯甲酰氯,dmap。其中所述化合物4∶无水四氢呋喃腈∶双(三甲基硅基)胺基锂∶对氟苯甲酰氯∶dmap的比例mmol∶ml∶mmol∶mmol∶mmol为17.0∶68∶18.7∶102.0∶17.0,反应液自然升温至过夜,使用饱和碳酸钾淬灭,乙酸乙酯萃取并旋蒸浓缩得到粗产物。通过柱色谱法纯化得到所需产物为白色固体,即为化合物methyl5-bromo-6-chloro-2-(4-fluorophenyl)furo[2,3-b]pyridine-3-carboxylate;
其中柱层析纯化使用石油醚,乙酸乙酯。石油醚∶乙酸乙酯的体积比100∶1~50∶1;
步骤f:化合物6的合成:
在氩气保护下,在干燥的双口瓶中依次加入化合物5,甲醇,氢氧化钠溶液(3n),在70℃下反应2h,其中所述化合物5∶甲醇∶氢氧化钠的比例mmol∶ml∶mmol为14.4∶72∶43.3,tlc监测反应完毕后使用盐酸(6n)调ph=4-5,反应瓶中有白色固体析出,过滤,真空烘箱烘干得到白色固体产物。即为化合物5-bromo-6-chloro-2-(4-fluorophenyl)furo[2,3-b]pyridine-3-carboxylicacid;
步骤g:bmt-052关键中间体i的合成:
在氩气保护下,在干燥的双口瓶中依次加入化合物6,四氢呋喃,甲胺盐酸盐,n,n-二异丙基乙胺,hbtu,其中所述化合物6∶无水四氢呋喃∶甲胺盐酸盐∶n,n-二异丙基乙胺∶hbtu的比例mmol∶ml∶mmol∶mmol∶mmol为13.6∶90∶67.8∶67.8∶20.1,反应体系在常温下过夜,tlc监测反应完毕后使用盐酸(1n)调ph为酸性,分离有机相,然后用乙酸乙酯萃取三次,合并有机相后分别用水,饱和食盐水洗涤,无水硫酸钠干燥,并旋蒸浓缩得到粗产物。通过柱色谱法纯化得到所需产物为白色固体。即为化合物5-bromo-6-chloro-2-(4-fluorophenyl)-n-methylfuro[2,3-b]pyridine-3-carboxamide。
其中柱层析纯化使用石油醚,乙酸乙酯。石油醚∶乙酸乙酯的体积比50∶1~10∶1;
本发明取得的优点和积极效果是:
1、本发明是以2-氯-3溴-5-甲基吡啶为原料经过溴化、氰基化、水解及氧化生成吡啶氮氧化物,在温和的条件下关环得到吡啶并呋喃母核。该方法工艺简单,反应条件温和,生产成本低,环境污染小,无过渡金属催化,整个合成不需要任何保护基,开拓了产率较高的制备bmt-052关键中间体的方法。
2、本发明作为一种全新的丙肝病毒ns5b聚合酶抑制剂bmt-052中间体的制备方法不仅产率高,环境污染小,而且工艺操作方便,产物易纯化和易放大生产,易于实现丙肝病毒ns5b聚合酶抑制剂bmt-052关键中间体的工业化生产。
附图说明
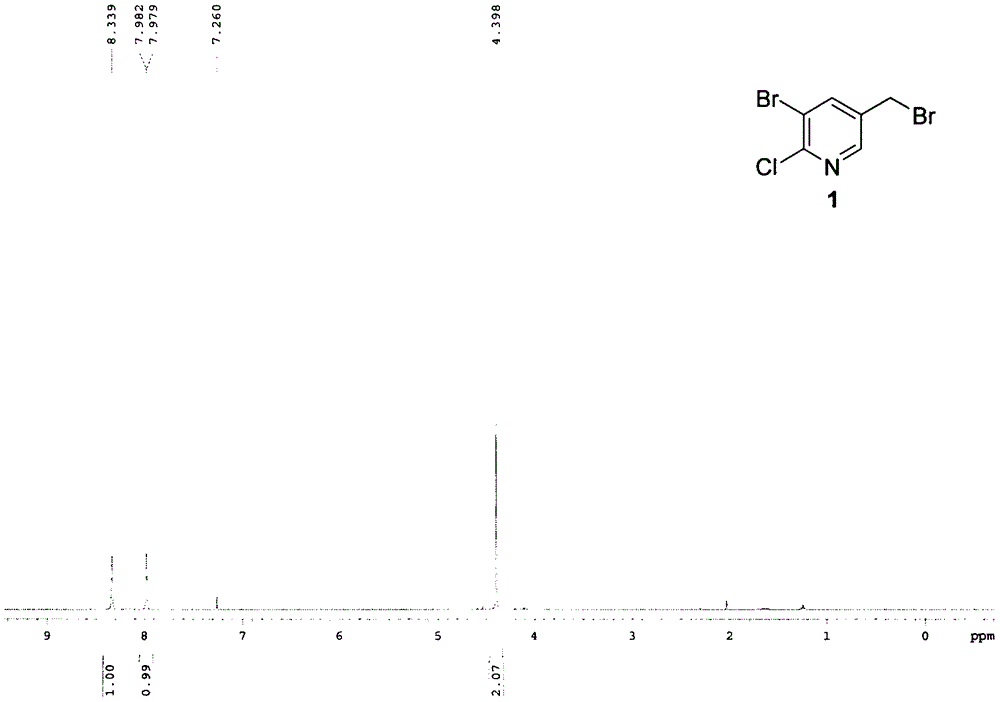
图1为本发明中化合物1在氘带三氯甲烷中的核磁共振氢谱图。
图2为本发明中化合物1在氘带三氯甲烷中的核磁共振碳谱图。
图3为本发明中化合物2在氘带三氯甲烷中的核磁共振氢谱图。
图4为本发明中化合物2在氘带三氯甲烷中的核磁共振碳谱图。
图5为本发明中化合物3在氘带三氯甲烷中的核磁共振氢谱图。
图6为本发明中化合物3在氘带三氯甲烷中的核磁共振碳谱图。
图7为本发明中化合物4在氘带二甲基亚砜中的核磁共振氢谱图。
图8为本发明中化合物4在氘带二甲基亚砜中的核磁共振碳谱图。
图9为本发明中化合物5在氘带三氯甲烷中的核磁共振氢谱图。
图10为本发明中化合物5在氘带三氯甲烷中的核磁共振碳谱图。
图11为本发明中化合物6在氘带三氯甲烷中的核磁共振氢谱图。
图12为本发明中化合物6在氘带三氯甲烷中的核磁共振碳谱图。
图13为本发明中化合物i在氘带三氯甲烷中的核磁共振氢谱图。
图14为本发明中化合物i在氘带三氯甲烷中的核磁共振碳谱图。
具体实施方式
为了理解本发明,下面结合实施例对本发明作进一步说明:下述实施例是说明性的,不是限定性的,不能以下述实施例来限定本发明的保护范围。
下面详细叙述本发明的实施例,需要说明的是,本实施例是叙述性的,不是限定性的,不能以此限定本发明的保护范围。
本发明中所使用的原料,如无特殊说明,均为常规的市售产品;本发明中所使用的方法,如无特殊说明,均为本领域的常规方法。
实施例1
一种丙肝病毒ns5b聚合酶抑制剂bmt-052中间体的高产率合成方法,包括以下步骤g:使化合物6生成bmt-052关键中间体i;
较优的所述化合物6生成bmt-052中间体是与甲胺盐酸盐,diea,hbtu反应下生成的。
较优的上述方法进一步包括步骤f:使化合物5反应生成化合物6;
较优的所述化合物5生成化合物6是与氢氧化钠溶液发生水解反应生成的。
较优的上述方法进一步包括步骤e:使化合物4反应生成化合物5;
较优的所述化合物4生成化合物5是与双(三甲基硅基)胺基锂,对氟苯甲酰氯,dmap反应生成的。
较优的上述方法进一步包括步骤d:使化合物3发生氧化反应反应生成化合物4;
较优的所述化合物3生成化合物4是与过氧化脲,三氟乙酸酐反应生成的。
较优的上述方法进一步包括步骤c:使化合物2反应生成化合物3;
较优的所述化合物2生成化合物3是与二氯亚砜发生反应生成的。
较优的上述方法进一步包括步骤b:使化合物1合成化合物2;
较优的所述化合物2生成化合物3是与氢氧化锂,tmscn发生反应生成的。
较优的上述方法进一步包括步骤a:使2-氯-3溴-5-甲基吡啶反应生成化合物1;
综上,本发明丙肝病毒ns5b聚合酶抑制剂bmt-052中间体i的高产率合成方法的合成路线如下:
实施例2
丙肝病毒ns5b聚合酶抑制剂bmt-052中间体的高产率合成方法,步骤如下:
步骤a:化合物1的合成:
在有磁力搅拌,氩气保护的250ml的双口瓶中加入2-氯-3溴-5-甲基吡啶(5g,24.22mmol)溶解在二氯乙烷中(70ml),随后加入n-溴代丁二酰亚胺(4.3g,36.32mmol)和过氧化苯甲酰(587mg,2.42mmol).反应液在93℃下反应6小时,tlc监测反应完毕,冷却至室温后用水淬灭(140ml)然后用乙酸乙酯萃取3次。合并有机相后用饱和食盐水洗涤,无水硫酸钠干燥。并旋蒸浓缩得到粗产物。通过柱色谱法纯化(石油醚∶乙酸乙酯=100∶1~50∶1)得到所需产物为白色固体(5.41g,79%收率),即为化合物1。1hnmr(400mhz,cdcl3)δ8.34(s,1h),7.98(d,j=1.2hz,1h),4.40(s,2h).13cnmr(100mhz,cdcl3)δ150.7,147.8,142.7,134.2,120.4,27.3.
步骤b:化合物2的合成:
在有磁力搅拌,氩气保护的250ml的双口瓶中加入化合物1(5.41g,19.13mmol)溶解在无水乙腈中(76ml),随后加入一水合氢氧化锂(963mg,22.96mmol)随后在0℃下滴加三甲基腈硅烷(2.28g,22.96mmol).反应液在常温下反应4小时,tlc监测反应完毕后用乙酸乙酯淬灭,加水萃取。然后用乙酸乙酯萃取3次。合并有机相后用饱和食盐水洗涤,无水硫酸钠干燥。并旋蒸浓缩得到粗产物。通过柱色谱法纯化(石油醚∶乙酸乙酯=20∶1~5∶1)得到所需产物为白色固体(4.18g,95%收率),即为化合物2。1hnmr(400mhz,cdcl3)δ8.30(d,j=2hz,1h),7.97(d,j=2hz,1h),3.77(s,2h).13cnmr(100mhz,cdcl3)δ151.0,147.0,141.7,126.4,120.9,116.0,20.2.
步骤c:化合物3的合成:
在有磁力搅拌,氩气保护的50ml的双口瓶中加入化合物2(4.18g,18.18mmol)溶解在甲醇中(15ml),将双口瓶在冰浴中降至0℃,随后缓慢滴加二氯亚砜(3.3ml,45.45mmol).反应液在常温下反应过夜,tlc监测反应完毕后使用旋转蒸发仪蒸干反应液,随后用饱和碳酸氢钠与二氯甲烷萃取。合并有机相后用饱和食盐水洗涤,无水硫酸钠干燥。并旋蒸浓缩得到粗产物。通过柱色谱法纯化(石油醚∶乙酸乙酯=30∶1~10∶1)得到所需产物为白色固体(4.47g,94%收率),即为化合物3。1hnmr(400mhz,cdcl3)δ8.23(d,j=2hz,1h),7.92(d,j=2hz,1h),3.73(s,3h),3.61(s,2h).13cnmr(100mhz,cdcl3)δ170.3,149.8,148.5,143.2,130.1,120.2,52.7,37.0.hrms(+esi-tof)m/z:[m+h]+calcdforc8h7no2clbr263.9421;found263.9411.
步骤d:化合物4的合成:
在有磁力搅拌,氩气保护的250ml的双口瓶中加入化合物3(4.47g,17.00mmol)溶解在二氯甲烷中(78ml),将双口瓶在冰浴中降至0℃,然后加入过氧化脲(7.53g,80.09mmol),随后缓慢滴加三氟乙酸酐(15.29g,72.70mmol)。反应液在常温下反应1h,tlc监测反应完毕后使用饱和亚硫酸钠淬灭,二氯甲烷萃取。合并有机相后用饱和食盐水洗涤,无水硫酸钠干燥。并旋蒸浓缩得到产物为白色固体,即为化合物4,可直接投入下步使用。1hnmr(400mhz,cdcl3)δ8.25(s,1h),7.45(s,1h),3.72(s,3h),3.54(s,2h).13cnmr(100mhz,cdcl3)δ169.3,142.4,139.8,130.9,130.5,121.2,52.8,37.1.hrms(+esi-tof)m/z:[m+na]+calcdforc8h7no3clbr301.9190;found301.9177.
步骤e:化合物5的合成:
在有磁力搅拌,氩气保护的250ml的双口瓶中加入上步所得化合物4(4.74g,17.00mmol)溶解在无水四氢呋喃中(68ml),将反应体系降至-78℃,然后滴加双(三甲基硅基)胺基锂(18.7ml,18.7mmol),在-78℃下反应2h,随后缓慢滴加对氟苯甲酰氯(16.17g,101.98mmol),dmap(2.08g,16.99mmol)。反应液自然升温至过夜,tlc监测反应完毕后使用饱和碳酸钾淬灭,乙酸乙酯萃取。合并有机相后用饱和食盐水洗涤,无水硫酸钠干燥。并旋蒸浓缩得到产物。通过柱色谱法纯化(石油醚∶乙酸乙酯=100∶1~50∶1)得到所需产物为白色固体(5.53g,85%收率),即为化合物5。1hnmr(400mhz,cdcl3)δ8.53(s,1h),8.14-8.18(m,2h),7.17-7.22(m,2h),3.97(s,3h).13cnmr(100mhz,cdcl3)δ164.7(d,j=253hz),163.0,160.8,157.6,145.8,137.2,132.2(d,j=9hz),124.1(d,j=3hz),120.2,116.1,115.9(d,j=22hz),107.2,52.4.hrms(+esi-tof)m/z:[m+h]+calcdforc15h8no3fclbr383.9433;found383.9434.
步骤f:化合物6的合成:
在有磁力搅拌的250ml的双口瓶中加入化合物5(5.53g,14.43mmol)溶解在甲醇(72ml)中,然后滴加氢氧化钠溶液(3n,43.29mmol),在70℃下反应2h,tlc监测反应完毕后使用盐酸(6n)调ph=4-5,反应瓶中有白色固体析出,过滤,真空烘箱烘干得到白色固体产物(5.09g,95%收率),即为化合物6。1hnmr(400mhz,dmso)δ13.71(s,1h),8.59(s,1h),8.15-8.18(m,2h),7.41-7.46(m,2h).13cnmr(100mhz,dmso)δ163.7(d,j=168hz),163.2,159.4,157.1,143.9,136.9,132.3(d,j=9hz),124.2,120.9,115.7(d,j=22hz),115.1,108.3.
步骤g:bmt-052关键中间体i的合成:
在有磁力搅拌,氩气保护的250ml的双口瓶中加入化合物6(5.0g,13.55mmol)溶解在四氢呋喃(90ml)中,然后依次加入甲胺盐酸盐(4.57g,67.75mmol),n,n-二异丙基乙胺(8.76g,67.75mmol),hbtu(7.61g,20.01mmol),反应体系在常温下过夜,tlc监测反应完毕后使用盐酸(1n)调ph为酸性,分离有机相,然后用乙酸乙酯萃取三次,合并有机相后分别用水,饱和食盐水洗涤,无水硫酸钠干燥,并旋蒸浓缩得到粗产物。通过柱色谱法纯化(石油醚∶乙酸乙酯=50∶1~10∶1)得到所需产物为白色固体(4.66g,90%收率),既得bmt-052关键中间体i。1hnmr(400mhz,cdcl3)δ8.47(s,1h),7.85-7.88(m,2h),7.19-7.24(m,2h),5.89(s,1h),2.98(d,j=4.8hz,3h).13cnmr(100mhz,cdcl3)δ164.4(d,j=252hz),162.8,157.9,154.8,145.7135.9,130.9(d,j=9hz),124.2,120.9(d,j=3hz),120.8,116.7(d,j=22hz),115.8,111.2。符合bmt-052关键中间体的结构特征。
- 还没有人留言评论。精彩留言会获得点赞!