一种烯烃对映选择性芳基炔基化的方法与流程
本发明涉及药物中间体合成技术领域,具体涉及一种烯烃对映选择性芳基炔基化的方法。
背景技术:
鞘氨醇-1-磷酸(sphingosine-1-phosphates1p)是一种具有重要生物学活性的溶血磷脂,其在细胞生存、生长、分化和迁移等多个生物学过程中起到关键作用的信号分子。鞘氨醇-1-磷酸以相对高的浓度储存在人血小板中,血小板中缺乏能够分解代谢鞘氨醇-1-磷酸的酶,鞘氨醇-1-磷酸在生理刺激(诸如生长因子、细胞因子以及受体激动剂和抗原)的激活下会直接释放到血流中。鞘氨醇-1-磷酸一方面对于血小板聚集和血栓形成起关键作用,但又会加重心血管疾病,另一方面在高密度脂蛋白(hdl)中相对高浓度的鞘氨醇-1-磷酸有益于动脉粥样化形成。例如,最近有研究表明鞘氨醇-1-磷酸与其它血溶性脂类,如鞘氨醇磷酰胆碱和溶血硫酸盐,通过刺激血管内皮产生强效抗动脉粥样硬化信号分子一氧化氮在hdl的临床效果上明显有益。此外,与溶血磷脂酸一样,鞘氨醇-1-磷酸也是某些类型癌症的标志物,并且有证据表明其在细胞分裂或增殖中的作用可对癌症发展具有影响。目前对鞘氨醇-1-磷酸的研究是医药研究人员的研究热点,并且已逐渐向鞘氨醇-1-磷酸在代谢中治疗性干预积极研究,达到通过对s1p调节减轻疾病和病症影响的目的。
国际专利wo2012074926a1新型氮杂环衍生物作为鞘氨醇-1-磷酸受体调节剂,公开了一组新型氮杂环丁烷衍生物,该新型氮杂环丁烷衍生物作为受体调节剂对鞘氨醇-1-磷酸有强效选择性,用于治疗多种与鞘氨醇-1-磷酸受体调节相关的疾病。该专利中公开了合成新型氮杂环丁烷衍生物的一重要中间体,该中间体的合成路线为:
上述合成步骤繁琐,产物经多步反应导致其产率低,手性纯度也不高,致使终产物新型氮杂环丁烷衍生物的产率低,导致新型氮杂环丁烷衍生物的生产成本高,不利于新型氮杂环丁烷衍生物作为鞘氨醇-1-磷酸手提调节剂的推广使用。
技术实现要素:
本发明目的在于提供一种烯烃对映选择性芳基炔基化的方法,在温和的反应条件下有手性选择性的一步反应制备1,2-二芳基-3-丁炔系列化合物,可通过本发明公开的合成路线制备鞘氨醇-1-磷酸受体调节剂新型氮杂环衍生物的重要中间体,反应收率高、手性纯度高。
为达成上述目的,本发明提出如下技术方案:一种烯烃对映选择性芳基炔基化的方法,末端烯烃a、末端炔烃b、二芳基碘鎓盐c在包含催化剂和配体的环境中发生对映选择性芳基炔基化反应生成1,2-二芳基-3-丁炔d;
具体合成路线为:
其中,末端烯烃a化学式中基团r1为芳基、杂环或脂肪烃基,末端炔烃b化学式中基团r2为芳基、杂环、环烷基、环烯基或tms,二芳基碘鎓盐c化学式中基团r3为芳基、杂环或-cf3。
进一步的,所述末端烯烃a、末端炔烃b和二芳基碘鎓盐c的当量比为1.0:2.0:1.25。
进一步的,所述配体为
进一步的,所述催化剂为铜催化剂,铜催化剂为cui、cubr、cucl、cuoac、cu(mecn)4pf6和cucn中的一种或多种。
进一步的,所述末端烯烃a、末端炔烃b、二芳基碘鎓盐c在包含催化剂和配体的溶剂中发生对映选择性芳基炔基化反应,所述溶剂为甲醇、甲苯、dme、乙腈、dmf、thf和二恶烷中的一种或多种。
进一步的,所述溶剂中溶解有碱式盐,所述碱式盐为cs2co3、na2co3、li2co3、k2co3、k3po4、nahco3、khco3或k2hpo4。
进一步的,所述溶剂中催化剂、配体和碱式盐的当量比为0.05:0.05:2.0。
由以上技术方案可知,本发明的技术方案提供的烯烃对映选择性芳基炔基化的方法,获得了如下有益效果:
本发明公开的烯烃对映选择性芳基炔基化的方法,烯烃、二芳基碘鎓盐和末端炔烃在铜催化剂作用下进行对映选择性芳基炔基化反应,三种反应物在温和的反应条件下发生偶联反应,反应物范围广,合成了综合价值高的1,2-二芳基-3-丁炔系列化合物,反应收率高、手性纯度也高。本发明通过偶联反应一步合成1,2-二芳基-3-丁炔系列化合物成功的关键在于在设定反应条件中应用手性bopa配体,本发明提出一种在高价铜催化剂作用下苯基自由基生成的新方法。
采用本发明公开的合成方法制备鞘氨醇-1-磷酸受体调节剂新型氮杂环衍生物的重要中间体,相较于现有技术的合成方案多步反应,本发明可直接一步反应,并且反应条件温和,收率高、手性纯度也高;应用于制备鞘氨醇-1-磷酸受体调节剂新型氮杂环衍生物,降低新型氮杂环衍生物的制造成本,有利于新型氮杂环衍生物的推广。
应当理解,前述构思以及在下面更加详细地描述的额外构思的所有组合只要在这样的构思不相互矛盾的情况下都可以被视为本公开的发明主题的一部分。
结合附图从下面的描述中可以更加全面地理解本发明教导的前述和其他方面、实施例和特征。本发明的其他附加方面例如示例性实施方式的特征和/或有益效果将在下面的描述中显见,或通过根据本发明教导的具体实施方式的实践中得知。
附图说明
附图不意在按比例绘制。在附图中,在各个图中示出的每个相同或近似相同的组成部分可以用相同的标号表示。为了清晰起见,在每个图中,并非每个组成部分均被标记。现在,将通过例子并参考附图来描述本发明的各个方面的实施例,其中:
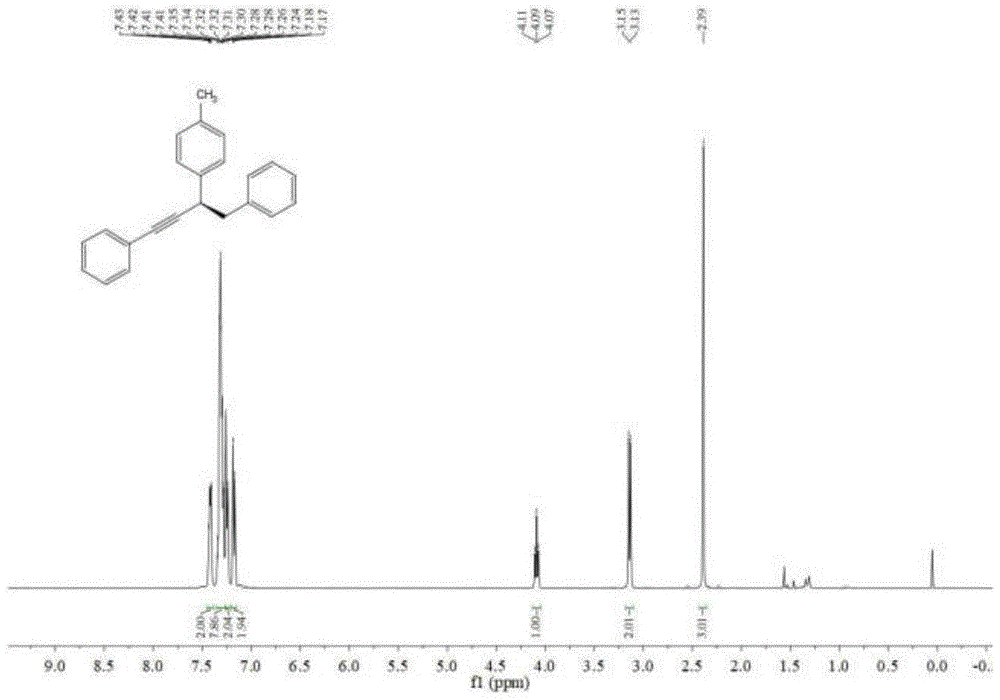
图1为本发明实施例1产物d1的核磁氢谱图;
图2为本发明实施例1产物d1的核磁碳谱图;
图3为本发明实施例1产物d1的高效液相色谱图;
图4为本发明实施例2产物d2的核磁氢谱图;
图5为本发明实施例2产物d2的核磁碳谱图;
图6为本发明实施例2产物d2的高效液相色谱图;
图7为本发明实施例中选择不同末端烯烃a对应的产物及其产率图;
图8为本发明实施例中选择不同末端炔烃b和二芳基碘鎓盐c对应的产物及其产率图;
图9为本发明合成路线的反应机理图;
图10为本发明优选的合成路线图。
具体实施方式
为了更了解本发明的技术内容,特举具体实施例并配合所附图式说明如下。
在本公开中参照附图来描述本发明的各方面,附图中示出了许多说明的实施例。本公开的实施例不定义包括本发明的所有方面。应当理解,上面介绍的多种构思和实施例,以及下面更加详细地描述的那些构思和实施方式可以以很多方式中任意一种来实施,这是因为本发明所公开的构思和实施例并不限于任何实施方式。另外,本发明公开的一些方面可以单独使用,或者与本发明公开的其他方面的任何适当组合来使用。
接于现有技术中制备鞘氨醇-1-磷酸受体调节剂新型氮杂环衍生物的合成中间体的合成路线,反应步骤多,反应条件不温和,产率低且产物的手性纯度不高的技术问题,本发明旨在提出一种烯烃对映选择性芳基炔基化的方法,在温和的反应条件下有手性选择性的一步反应制备1,2-二芳基-3-丁炔系列化合物,反应步骤简单,反应收率及产物的手性纯度高,当本发明的合成方法应用于制备鞘氨醇-1-磷酸受体调节剂新型氮杂环衍生物的重要中间体时,有利于鞘氨醇-1-磷酸受体调节剂新型氮杂环衍生物制备及推广应用。
本发明公开的烯烃对映选择性芳基炔基化的方法,具体为:末端烯烃a、末端炔烃b、二芳基碘鎓盐c在包含催化剂和配体的环境中发生对映选择性芳基炔基化反应生成1,2-二芳基-3-丁炔d;
具体合成路线为:
其中,末端烯烃a化学式中基团r1为芳基、杂环或脂肪烃基,末端炔烃b化学式中基团r2为芳基、杂环、环烷基、环烯基或tms(四甲基硅烷),二芳基碘鎓盐c化学式中基团r3为芳基、杂环或-cf3。催化剂选用铜催化剂,铜催化剂为cui、cubr、cucl、cuoac、cu(mecn)4pf6和cucn中的一种或多种;所述配体为
具体而言,末端烯烃a、末端炔烃b、二芳基碘鎓盐c在包含催化剂和配体的溶剂中发生对映选择性芳基炔基化反应,溶剂为甲醇、甲苯、dme、乙腈、dmf、thf和二恶烷中的一种或多种;溶剂中溶解有碱式盐,碱式盐使得溶剂呈碱性,所述碱式盐为cs2co3、na2co3、li2co3、k3co3、k3po4、nahco3、khco3和k2hpo4中的一种或多种。
本发明在具体实施时,反应条件温和,在室温下即可进行反映;另外,为确保反应物在反应过程中不与外界环境元素反应,反应在无水无氧条件下操作,本发明公开的对映选择性芳基炔基化反应在惰性气体保护体系下进行反应,如氮气体系、氩气体系,说明书实施例中均直接采用氮气体系。
本发明烯烃对映选择性芳基炔基化的方法的合成路线具体操作过程为:在干燥的密封管中,将配体(0.05当量)、碱式盐(2.0当量)和铜催化剂(0.05当量)在室温下完全溶解于溶剂中;在氮气气氛下,将上述混合物搅拌一段时间,然后依次加入末端烯烃(1.0当量)、末端炔烃(2.0当量)和二芳基碘鎓盐(1.25当量),反应物添加完成后密封管开口密封;混合物在室温下搅拌反应24小时,通过tlc板监控反应直至反应完成,产物通过硅胶柱色谱法纯化,用石油醚和乙酸乙酯作为洗脱剂。
下面结合具体实施例,对本发明的烯烃对映选择性芳基炔基化的方法,作进一步具体介绍。
实施例1
在干燥的密封管中,将配体tbu-bopa(0.037mmol、0.05当量)、碱式盐k2co3(0.4mmol、2.0当量)和铜催化剂(cu(mecn)4pf6、0.05当量)在室温下溶解于无水乙腈(2.0ml)中;在氮气气氛下,将上述混合物搅拌10min,然后依次加入对甲基苯乙烯(0.2mmol、1.0当量)、苯乙炔(0.4mmol、2.0当量)和二苯基碘鎓六氟磷酸盐(0.25mmol、1.25当量),反应物添加完成后采用铁氟龙隔膜对密封管开口密封;混合物在室温下搅拌反应24小时,通过tlc板监控反应直至反应完成,产物通过硅胶柱色谱法纯化,用石油醚和乙酸乙酯作为洗脱剂;得到无色油状产物d1,46.2mg,0.16mmol,收率78%,对映体过量值86%。
产物d1的核磁图谱及高效液相色谱图如附图1至图3所示,1hnmr(400mhz,cdcl3)δ7.45–7.38(m,2h),7.35–7.27(m,8h),7.25(d,j=7.7hz,2h),7.18(d,j=7.7hz,2h),4.09(t,j=7.3hz,1h),3.14(d,j=7.3hz,2h),2.39(s,3h)。
13cnmr(100mhz,cdcl3)δ139.0,138.3,136.4,131.5,129.5,129.1,128.1,128.0,127.7,127.5,126.4,123.7,91.2,84.1,45.1,40.4,21.1.ir(neat)cm-1
hplc(chiralcelod-hcolumn,hexanes:i-proh=100:0,0.8ml/min,210nm),tminor=25.9min,tmajor=29.4min,ee=86%.[α]d25=5.4,(c=0.47,chcl3)。
实施例2
与实施例1不同的是反应物a更换为4-乙酰氧基苯乙烯(0.2mmol、1.0当量),其它条件不变,得到无色油状产物d2,56.4mg,0.17mmol,收率83%,对映体过量值89%。
产物d2的核磁图谱及高效液相色谱图如附图4至图6所示:1hnmr(400mhz,cdcl3)δ7.90(d,j=7.9hz,2h),7.35–7.27(m,4h),7.23–7.19(m,3h),7.18–7.12(m,3h),7.05(d,j=6.9hz,2h),4.05(t,j=7.1hz,1h),3.82(s,3h),3.10–2.97(m,2h)。
13cnmr(100mhz,cdcl3)δ166.9,146.4,138.2,131.5,129.7,129.5,128.8,128.2,128.1,128.0,127.8,126.6,123.3,90.0,84.7,52.0,44.7,40.7.ir(neat)cm-1
hplc(chiralcelod-hcolumn,hexanes:i-proh=98:2,0.8ml/min,210nm),tminor=8.9min,tmajor=9.9min,ee=89%.[α]d25=8.1,(c=0.15,chcl3)。
实施例3
与实施例1不同的是反应物a更换为对三氟甲基苯乙烯(0.2mmol、1.0当量),其它条件不变,得到无色油状产物d3,55.3mg,0.16mmol,收率86%,对映体过量值86%。
产物d3的核磁图谱及高效液相色谱数据结果如下:1hnmr(400mhz,cdcl3)δ7.56(d,j=7.9hz,2h),7.44(d,j=8.0hz,2h),7.41–7.35(m,2h),7.32–7.19(m,6h),7.14(d,j=7.1hz,2h),4.13(t,j=7.2hz,1h),3.19–3.05(m,2h)。
13cnmr(100mhz,cdcl3)δ140.2,138.1,131.5,129.5,129.2(q,j=32.3hz),128.3,128.1,128.1,128.0,126.7,125.4(q,j=3.0hz),124.2(q,j=270.1hz),123.2,89.9,84.8,44.8,40.1.19fnmr(376mhz,cdcl3)δ-62.2(s,3f).ir(neat)cm-1
hplc(chiralcelod-hcolumn,hexanes:i-proh=99.2:0.8,0.35ml/min,210nm),tminor=15.2min,tmajor=15.9min,ee=86%.[α]d25=4.9,(c=0.28,chcl3)。
实施例4
与实施例1不同的是反应物a更换为对苯乙烯(0.2mmol、1.0当量),其它条件不变,得到无色油状产物d4,40.6mg,0.14mmol,收率72%,对映体过量值87%。
产物d4的核磁图谱及高效液相色谱数据结果如下:1hnmr(400mhz,cdcl3)δ7.49–7.42(m,4h),7.41–7.28(m,9h),7.26(d,j=7.2hz,2h),4.15(t,j=7.2hz,1h),3.19(d,j=7.2hz,2h)。
13cnmr(100mhz,cdcl3)δ141.2,138.8,131.5,129.5,128.4,128.1,128.0,127.8,127.7,126.8,126.4,123.6,90.9,84.3,45.1,40.8.ir(neat)cm-1
hplc(chiralcelod-hcolumn,hexanes:i-proh=100:0,0.8ml/min,210nm),tminor=34.0min,tmajor=30.7min,ee=87%.[α]d25=103.8(c=0.05,chcl3)。
实施例5
与实施例1不同的是反应物a更换为对苯基苯乙烯(0.2mmol、1.0当量),其它条件不变,得到无色油状产物d5,53.7mg,0.15mmol,收率75%,对映体过量值89%。
产物d5的核磁图谱及高效液相色谱数据结果如下:1hnmr(400mhz,cdcl3)δ7.56(dd,j=16.7,7.7hz,4h),7.45–7.37(m,5h),7.36–7.15(m,9h),4.11(t,j=7.2hz,1h),3.14(d,j=7.2hz,2h)。
13cnmr(100mhz,cdcl3)δ140.8,140.4,139.7,138.8,131.5,129.5,128.7,128.2,128.1,128.0,127.8,127.2,127.1,127.0,126.5,123.6,90.8,84.4,45.0,40.5.ir(neat)cm-1
hplc(chiralcelod-hcolumn,hexanes:i-proh=99.5:0.5,0.8ml/min,210nm),tminor=13.7min,tmajor=15.7min,ee=89%.[α]d25=7.0,(c=0.33,chcl3)。
综上实施例1至实施例5所示的调节反应物a末端烯烃的结构,还包括其它末端烯烃,含有不同基团r1的末端烯烃与末端炔烃、二芳基碘鎓盐反应后产物、产率及对映体过量值均记载在图7所示的结构上。
本发明进一步公开了具有不同基团r2对应的反应物b、不同基团r3对应的反应物c与末端烯烃反应生成的不同产物、产率及对映体过量值,具体结果如附图8所示。
为进一步研究设定条件对本发明制备1,2-二芳基-3-丁炔系列化合物的影响,实施例6、实施例7、实施例8和实施例9分别研究了不同配体、不同铜催化剂、不同溶剂和不同碱性盐对产物收率和对映体过量值的影响。
实施例6:配体筛选
选取对甲基苯乙烯、苯乙炔和二苯基碘鎓六氟磷酸盐反应,反应式如下,选择配体l1至l11,以及2,2,2-三联吡啶共12种配体进行实验,验证配体对产物d1产率和对映体过量值的影响,结果如表1所示。
结合表1所示的产率及对映体过量值,配体l9对应的产物产率及对映体过量值最高,映体过量值高表明产物的手性纯度高,选定配体l9作为本次验证效果最佳的配体,并以配体l9选定较佳的铜催化剂。
表1.配体筛选
实施例7:催化剂筛选
选取对甲基苯乙烯、苯乙炔和二苯基碘鎓六氟磷酸盐反应,反应式如下,选择了cui、cubr、cucl、cuoac、cu(mecn)4pf6和cucn共6种铜催化剂实验,验证催化剂对产物d1产率和对映体过量值的影响,结果如表2所示。
表2.催化剂筛选
综合表2所示的产率及对映体过量值,具有较佳催化效果的铜催化剂为cu(mecn)4pf6,以配体l9、催化剂cu(mecn)4pf6进一步进行溶剂筛选。
实施例8:溶剂筛选
选取对甲基苯乙烯、苯乙炔和二苯基碘鎓六氟磷酸盐反应,反应式如下,选择了甲醇、甲苯、dme、乙腈、dmf、thf和二恶烷共7种溶剂实验,验证溶剂对产物d1产率和对映体过量值的影响,结果如表3所示。
表3.溶剂筛选
由表3中的数据结果可知,乙腈作为溶剂时,获得产物产率及对映体过量值均较高,再以配体l9、催化剂cu(mecn)4pf6、溶剂乙腈进一步进行碱式盐筛选。
实施例8:碱式盐筛选
选取对甲基苯乙烯、苯乙炔和二苯基碘鎓六氟磷酸盐反应,反应式如下,选择了cs2co3、na2co3、li2co3、k2co3、k3po4、nahco3、khco3和k2hpo4共8种碱式盐实验,验证碱式盐对产物d1产率和对映体过量值的影响,结果如表4所示。
表4.碱式盐筛选
由表4所示的碱式盐筛选数据,表明k2co3对本发明的烯烃对映选择性芳基炔基化具有较佳的促进效果。
综上实施例1至实施例8所述,获得本发明的反应机理如图9所示。
本发明的反应机理如图9所示,cu(i)x在碱性环境中与配体ln反应生成络合物cu(i)ln。随后在碱性环境中,络合物cu(i)ln与炔烃作用生成[lncu(i)(c≡cr)]-k+(m),[lncu(i)(c≡cr)]-k+(m)被二芳基碘鎓盐氧化,得到cu(ii)粒子,lncu(ii)(c≡cr)(n),构成芳基。综上所述,本发明烯烃对映选择性芳基炔基化的方法,优选的合成路线如图10所示。
本发明公开的烯烃对映选择性芳基炔基化的方法,对于国际专利wo2012074926a1公开的新型氮杂环衍生物作为鞘氨醇-1-磷酸受体调节剂中合成新型氮杂环衍生物的重要中间体的过程步骤可直接修改为如下合成路线:
该合成路线相较于该国际专利公开的反应步骤由现有的5步反应直接一步完成,反应条件温和,产物的收率高,对映体过量值也高。
虽然本发明已以较佳实施例揭露如上,然其并非用以限定本发明。本发明所属技术领域中具有通常知识者,在不脱离本发明的精神和范围内,当可作各种的更动与润饰。因此,本发明的保护范围当视权利要求书所界定者为准。
- 还没有人留言评论。精彩留言会获得点赞!