一种药物中间体的手性提纯方法与流程
本发明属于手性提纯技术领域,具体涉及一种药物中间体的手性提纯方法。
背景技术:
anacetrapib(安塞曲匹)用于遭受心血管病的患者,以降低冠心病发作、死亡的风险。anacetrapib是一种胆固醇酯转运蛋白抑制剂,可增加高密度脂蛋白(hdl)胆固醇并降低低密度脂蛋白(ldl)胆固醇水平,且不会对血压造成影响,因临床效果较好而有着潜在广阔的市场需求和社会价值。研究者发现,将1623例病人接受了随机分组。在24周时,anacetrapib组的ldl胆固醇水平已从81mg/dl(2.1mmol/l)降至45mg/dl(1.2mmol/l),相比之下,安慰剂组从82mg/dl(2.1mmol/l)降至77mg/dl(2.0mmol/l)(p<0.001),相对于anacetrapib组的降幅比安慰剂组观察到的多39.8%的显著效果,anacetrapib的结构式如式i所示:
anacetrapib由其重要的药物中间体a4合成,但由于药物中间体a4具有的化学结构和其所含对映体杂质的化学结构决定,其很难通过化学反应的方法进行拆分,例如加入拆分剂进行拆分,而通过手性柱进行拆分,损失很大,收率很低,尤其当其光学纯度本身较高时更是难以进行手性拆分。
因此,如何进一步提高药物中间体a4的光学纯度是本领域目前研究的重点。
技术实现要素:
本发明的目的在于提供一种药物中间体的手性提纯方法,所述手性提纯方法能够使得a4的光学纯度达到99.90-99.99%,并且收率较高,可达到90%以上。
为达此目的,本发明采用以下技术方案:
本发明提供一种药物中间体的手性提纯方法,所述药物中间体的手性提纯方法包括以下步骤:
(1)将低光学纯度的式a4所示化合物加入到溶剂a中,混合搅拌,过滤,得到滤液;
(2)将步骤(1)得到的滤液进行一次浓缩后,加入溶剂b溶解,再进行二次浓缩,最后再加入溶剂b混合搅拌,过滤,得到高光学纯度的式a4所示化合物;
所述式a4所示化合物的结构式如下所示:
所述低光学纯度的式a4所示化合物中存在少量其对映体杂质化合物a4,a4的结构式如下所示:
本发明能够通过溶剂提纯的原因如下所述:当一分子a4所示化合物与一分子a4所示化合物通过氢键结合形成8元环结构c1,原分子中除5元杂环可与之共面,其他原子处在面的两侧,一分子内裸露的甲基和双三氟甲基苯基,与另一分子裸露的甲基和双三氟甲基苯基的斥力小,可以形成稳定的二聚体,使其在溶剂a中的溶解度变小,容易以a4:a4摩尔质量比为1:1的形式从步骤(1)中的溶剂a中析出,即步骤(1)过滤得到的滤饼。
而两分子a4通过氢键结合形成8元环结构c2,原分子中除5元杂环可与之共面,其他原子处在面的上方,一分子内裸露的甲基和双三氟甲基苯基,与另一分子裸露的甲基和双三氟甲基苯基的斥力较大,不能形成稳定的二聚体,容易以单分子的形式被溶剂a溶解,即完全溶解在步骤(1)得到的滤液中。
其中,低光学纯度的式a4所示化合物由如下路线合成:
该路线使用cbz-l-丙氨酸为起始原料与二甲羟安缩合得到a1,经三氟甲基溴苯反应得到a2(油状物),不需纯化即可投下一步,避免纯化导致收率低(60%收率),经异丙醇铝还原后得到a3,避免其他路线使用价格较贵的二甲基苯基硅烷,后经氢氧化钾处理,即可得到a4。因此该路线使用原料较便宜,合成路线简单,总收率较高。
优选地,步骤(1)中所述低光学纯度的式a4所示化合物的光学纯度为86.00-99.50%,例如可以是86.00%、87.00%、88.00%、89.00%、90.00%、91.00%、92.00%、93.00%、94.00%、95.00%、96.00%、97.00%、98.00%、99.00%、99.50%。
优选地,步骤(1)中所述溶剂a包括二氯甲烷、氯仿、甲醇、乙醇或乙酸乙酯中的任意一种或至少两种的混合物,优选为二氯甲烷。
优选地,步骤(1)中所述混合搅拌的温度为10-11℃,例如可以是10℃、10.1℃、10.2℃、10.3℃、10.4℃、10.5℃、10.6℃、10.7℃、10.8℃、10.9℃、11℃。
优选地,步骤(1)中所述混合搅拌的时间为0.4-0.6h,例如可以是0.4h、0.45h、0.5h、0.55h、0.6h,优选为0.5h。
优选地,步骤(2)中所述高光学纯度的式a4所示化合物的光学纯度为99.90-99.99%,例如可以是99.90%、99.91%、99.92%、99.93%、99.94%、99.95%、99.96%、99.97%、99.98%、99.99%。
优选地,步骤(2)中所述溶剂b为正庚烷、正己烷或环己烷中的任意一种或至少两种的混合物,优选为正庚烷。
优选地,步骤(2)中所述一次浓缩的温度为30-40℃,例如可以是30℃、32℃、34℃、36℃、38℃、40℃。
优选地,步骤(2)中所述二次浓缩的温度为30-40℃,例如可以是30℃、32℃、34℃、36℃、38℃、40℃。
优选地,步骤(2)中所述混合搅拌的温度为5-10℃,例如可以是5℃、6℃、7℃、8℃、9℃、10℃。
优选地,步骤(2)中所述混合搅拌的时间为0.4-0.6h,例如可以是0.4h、0.45h、0.5h、0.55h、0.6h,优选为0.5h。
优选地,所述药物中间体的手性提纯方法包括以下步骤:
(1)将光学纯度为86.00-99.50%的式a4所示化合物加入到溶剂a中,10-11℃下混合搅拌0.4-0.6h,过滤,得到滤液;
(2)将步骤(1)得到的滤液在30-40℃下进行一次浓缩后,加入溶剂b溶解,再在30-40℃下进行二次浓缩,最后再加入溶剂b,在5-10℃下混合搅拌0.4-0.6h,过滤,得到光学纯度为99.90-99.99%的式a4所示化合物。
相对于现有技术,本发明具有以下有益效果:
本发明所述药物中间体的手性提纯方法,采用廉价的溶剂即能进行手性提纯,条件温和,操作简单,能够使得式a4所示化合物的光学纯度达到99.90-99.99%,且收率较高,可达到90%以上。
附图说明
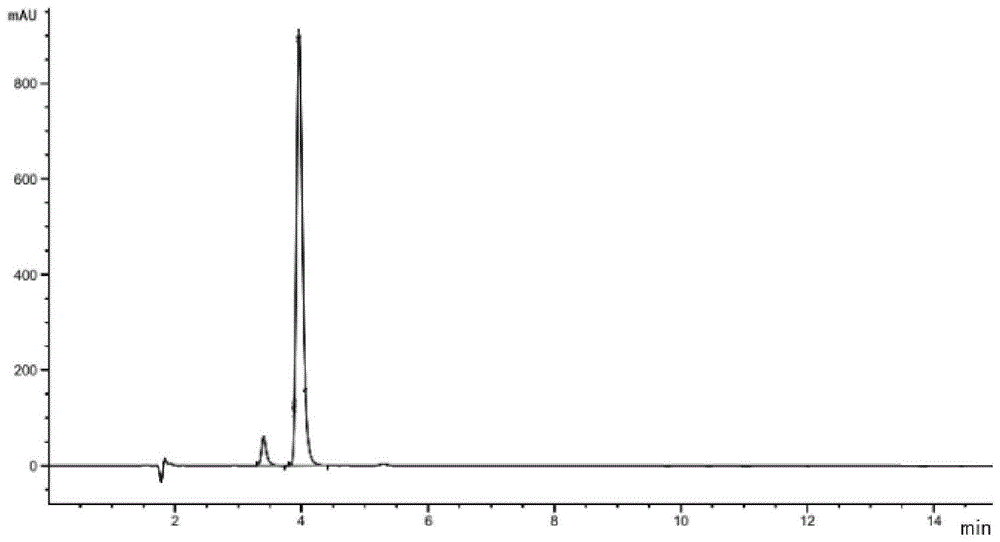
图1为实施例4低光学纯度的式a4所示化合物的hplc检测结果图。
图2为实施例4高光学纯度的式a4所示化合物的hplc检测结果图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
本实施例提供一种药物中间体的手性提纯方法,所述药物中间体的手性提纯方法包括以下步骤:
(1)在1l的三口烧瓶中,将光学纯度为94.8%的式a4所示化合物(66.8g)加入到二氯甲烷(330ml)中,在10℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为51:49的混合物),收集滤液;
(2)将步骤(1)得到的滤液在35℃下浓缩后,加入正庚烷(180ml)浓缩,再加入正庚烷(180ml)浓缩,最后再加入正庚烷(200ml)在5℃下混合搅拌0.5h,过滤,滤饼烘干得56.2g,收率84.1%,得到光学纯度为99.93%的式a4所示化合物。
本实施例提供的光学纯度为94.8%的式a4所示化合物合成路线为:
(1)中间体a1的合成
向5l三口瓶中加入四氢呋喃1100g,cbz-l-丙氨酸289.5g,hobt211.2g,二甲羟胺盐酸盐150.9g,dmap15.9g,二氯甲烷300g,10-20℃下搅拌10min,滴加三乙胺330g,控温20℃以内,滴完后,搅拌30min。加入edc盐酸盐300g,控温20-25℃,保温8h。向体系中加入乙酸乙酯1200g,控温25℃以内,滴加3n盐酸1440g,搅拌后分液,有机相用1800g碳酸氢钠饱和溶液洗,再用450g饱和氯化钠溶液洗,分液。有机相浓缩干,用1500g正庚烷打浆,抽滤。滤饼烘干得a1293.3g,收率85%,化学纯度99.2%,手性纯度99.7%。
(2)中间体a2的合成
向5l反应釜中加入a1293.3g,四氢呋喃600g,3,5-双(三氟甲基)溴苯400g,氮气保护下,降温至-10℃,滴加2m异丙基氯化镁四氢呋喃溶液1500g,控温-5~0℃,滴完后反应2h。在0-10℃下向体系滴加3n盐酸900g,加入乙酸乙酯820g,搅拌30min,分层,有机相用375g饱和食盐水洗,浓缩,再用甲苯150g*2带2次,得a2粗品550g黄色油状物。
(3)中间体a3的合成
向a2粗品中加入甲苯900g,异丙醇480g,异丙醇铝72kg,氮气保护下,升温至50℃,保温16h。把体系降温至10℃以内,加入甲苯1800g,1n盐酸1200g,搅拌30min,分液。有机相浓缩干,加入正庚烷2000g,打浆4h,抽滤。烘干得a3301.2g,a2、a3两步收率64.5%,化学纯度99.2%,手性纯度94.4%。
(4)光学纯度为94.8%的中间体a4的合成
向5l反应釜中加入甲苯3000g,异丙醇300g,a3300g,降温至10℃以内,加入氢氧化钾80g,升温至20℃,保温2h。体系降温至10℃以内,滴加1900g的1n盐酸,滴完后,搅拌30min,分液。有机相浓缩干,加入2200g正庚烷,控温10-20℃,打浆4h,过滤。滤饼烘干,得a4180g,收率81%,化学纯度99.5%,手性纯度94.8%。
实施例2
本实施例提供一种药物中间体的手性提纯方法,所述药物中间体的手性提纯方法包括以下步骤:
(1)在1l的三口烧瓶中,将光学纯度为93.5%的式a4所示化合物(60g)加入到甲醇(150ml)中,在10℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为52.7:47.3的混合物),收集滤液;
(2)将步骤(1)得到的滤液在30℃下浓缩后,加入正庚烷(180ml)浓缩,再加入正庚烷(180ml)浓缩,最后再加入正庚烷(200ml),在5℃下混合搅拌0.5h,过滤,滤饼烘干得47.6g,收率79.3%,得到光学纯度为99.90%的式a4所示化合物。
本实施例提供的光学纯度为93.5%的式a4所示化合物合成路线为:
(1)中间体a1的合成
向5l三口瓶中加入四氢呋喃760g,cbz-l-丙氨酸193g,hobt140.8g,二甲羟胺盐酸盐100.6g,dmap10.6g,二氯甲烷200g,10-20℃下搅拌10min,滴加三乙胺220g,控温20℃以内,滴完后,搅拌30min。加入edc盐酸盐200g,控温20-25℃,保温8h。向体系中加入乙酸乙酯800g,控温25℃以内,滴加3n盐酸960g,搅拌后分液,有机相用1240g碳酸氢钠饱和溶液洗,再用300g饱和氯化钠溶液洗,分液。有机相浓缩干,用1000g正庚烷打浆,抽滤。滤饼烘干得a1202g,收率87.8%,化学纯度99.1%,手性纯度99.9%。
(2)中间体a2的合成
向5l三口瓶中加入a1197.6g,四氢呋喃400g,3,5-双(三氟甲基)溴苯274g,氮气保护下,降温至-10℃,滴加2m异丙基氯化镁四氢呋喃溶液1000g,控温-5~0℃,滴完后反应2h。在0-10℃下向体系滴加3n盐酸640g,加入乙酸乙酯560g,搅拌30min,分层,有机相用260g饱和食盐水洗,浓缩,再用甲苯120g*2带2次,得a2粗品340g黄色油状物。
(3)中间体a3的合成
向a2粗品中加入甲苯600g,异丙醇320g,异丙醇铝48.8g,氮气保护下,升温至50℃,保温16h。把体系降温至10℃以内,加入甲苯1200g,1n盐酸800g,搅拌30min,分液。有机相浓缩干,加入正庚烷1240g,打浆4h,抽滤。烘干得a3210.7g,a2、a3两步收率67.4%,化学纯度99.4%,手性纯度93.1%。
(4)光学纯度为93.5%的中间体a4的合成
向5l三口瓶中加入甲苯2000g,异丙醇200g,a3200g,降温至10℃以内,加入氢氧化钾57.5g,升温至20℃,保温2h。体系降温至10℃以内,滴加760g的1n盐酸,滴完后,搅拌30min,分液。有机相浓缩干,加入860g正庚烷,控温10-20℃,打浆4h,过滤。滤饼烘干,得a4126.6g,收率85.2%,化学纯度99.5%,手性纯度93.5%。
实施例3
本实施例提供一种药物中间体的手性提纯方法,所述药物中间体的手性提纯方法包括以下步骤:
(1)在1l的三口烧瓶中,将光学纯度为93.5%的式a4所示化合物(60g)加入到乙醇(150ml)中,在11℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为51.8:48.2的混合物),收集滤液;
(2)将步骤(1)得到的滤液在40℃下浓缩后,加入正庚烷(180ml)浓缩,再加入正庚烷(180ml)浓缩,最后再加入正庚烷(200ml)在10℃下混合搅拌0.5h,滤饼烘干得48.8g,收率81.3%,得到光学纯度为99.90%的式a4所示化合物。
本实施例提供的光学纯度为93.5%的式a4所示化合物合成路线为:
(1)中间体a1的合成
向5l三口瓶中加入四氢呋喃760g,cbz-l-丙氨酸193g,hobt140.8g,二甲羟胺盐酸盐100.6g,dmap10.6g,二氯甲烷200g,10-20℃下搅拌10min,滴加三乙胺220g,控温20℃以内,滴完后,搅拌30min。加入edc盐酸盐200g,控温20-25℃,保温8h。向体系中加入乙酸乙酯800g,控温25℃以内,滴加3n盐酸960g,搅拌后分液,有机相用1240g碳酸氢钠饱和溶液洗,再用300g饱和氯化钠溶液洗,分液。有机相浓缩干,用1000g正庚烷打浆,抽滤。滤饼烘干得a1202g,收率87.8%,化学纯度99.1%,手性纯度99.9%。
(2)中间体a2的合成
向5l三口瓶中加入a1197.6g,四氢呋喃400g,3,5-双(三氟甲基)溴苯274g,氮气保护下,降温至-10℃,滴加2m异丙基氯化镁四氢呋喃溶液1000g,控温-5~0℃,滴完后反应2h。在0-10℃下向体系滴加3n盐酸640g,加入乙酸乙酯560g,搅拌30min,分层,有机相用260g饱和食盐水洗,浓缩,再用甲苯120g*2带2次,得a2粗品340g黄色油状物。
(3)中间体a3的合成
向a2粗品中加入甲苯600g,异丙醇320g,异丙醇铝48.8g,氮气保护下,升温至50℃,保温16h。把体系降温至10℃以内,加入甲苯1200g,1n盐酸800g,搅拌30min,分液。有机相浓缩干,加入正庚烷1240g,打浆4h,抽滤。烘干得a3210.7g,a2、a3两步收率67.4%,化学纯度99.4%,手性纯度93.1%。
(4)光学纯度为93.5%的中间体a4的合成
向5l三口瓶中加入甲苯2000g,异丙醇200g,a3200g,降温至10℃以内,加入氢氧化钾57.5g,升温至20℃,保温2h。体系降温至10℃以内,滴加760g的1n盐酸,滴完后,搅拌30min,分液。有机相浓缩干,加入860g正庚烷,控温10-20℃,打浆4h,过滤。滤饼烘干,得a4126.6g,收率85.2%,化学纯度99.5%,手性纯度93.5%。
实施例4
本实施例提供一种药物中间体的手性提纯方法,所述药物中间体的手性提纯方法包括以下步骤:
(1)在1l的三口烧瓶中,将光学纯度为94.77%的式a4所示化合物(100g)加入到甲醇(570ml)中,在10℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为50.5:49.5的混合物),收集滤液;
(2)将步骤(1)得到的滤液在35℃下浓缩后,加入正庚烷(300ml)浓缩,再加入正庚烷(300ml)浓缩,最后再加入正庚烷(200ml)在10℃下混合搅拌0.5h,滤饼烘干得47.6g,收率79.3%,得到光学纯度为99.91%的式a4所示化合物。
其中,图1为实施例4低光学纯度的式a4所示化合物的hplc检测结果图。通过积分面积可得低光学纯度的式a4所示化合物的光学纯度为94.77%。
图2为实施例4高光学纯度的式a4所示化合物的hplc检测结果图。通过积分面积可得高光学纯度的式a4所示化合物的光学纯度为99.91%。
本实施例提供的光学纯度为94.77%的式a4所示化合物合成路线为:
(1)中间体a1的合成
向5l三口瓶中加入四氢呋喃1100g,cbz-l-丙氨酸289.5g,hobt211.2g,二甲羟胺盐酸盐150.9g,dmap15.9g,二氯甲烷300g,10-20℃下搅拌10min,滴加三乙胺330g,控温20℃以内,滴完后,搅拌30min。加入edc盐酸盐300g,控温20-25℃,保温8h。向体系中加入乙酸乙酯1200g,控温25℃以内,滴加3n盐酸1440g,搅拌后分液,有机相用1800g碳酸氢钠饱和溶液洗,再用450g饱和氯化钠溶液洗,分液。有机相浓缩干,用1500g正庚烷打浆,抽滤。滤饼烘干得a1293.3g,收率85%,化学纯度99.2%,手性纯度99.7%。
(2)中间体a2的合成
向5l反应釜中加入a1293.3g,四氢呋喃600g,3,5-双(三氟甲基)溴苯400g,氮气保护下,降温至-10℃,滴加2m异丙基氯化镁四氢呋喃溶液1500g,控温-5~0℃,滴完后反应2h。在0-10℃下向体系滴加3n盐酸900g,加入乙酸乙酯820g,搅拌30min,分层,有机相用375g饱和食盐水洗,浓缩,再用甲苯150g*2带2次,得a2粗品550g黄色油状物。
(3)中间体a3的合成
向a2粗品中加入甲苯900g,异丙醇480g,异丙醇铝72kg,氮气保护下,升温至50℃,保温16h。把体系降温至10℃以内,加入甲苯1800g,1n盐酸1200g,搅拌30min,分液。有机相浓缩干,加入正庚烷2000g,打浆4h,抽滤。烘干得a3301.2g,a2、a3两步收率64.5%,化学纯度99.2%,手性纯度94.4%。
(4)光学纯度为94.8%的中间体a4的合成
向5l反应釜中加入甲苯3000g,异丙醇300g,a3300g,降温至10℃以内,加入氢氧化钾80g,升温至20℃,保温2h。体系降温至10℃以内,滴加1900g的1n盐酸,滴完后,搅拌30min,分液。有机相浓缩干,加入2200g正庚烷,控温10-20℃,打浆4h,过滤。滤饼烘干,得a4180g,收率80.6%,化学纯度99.5%,手性纯度94.77%。
实施例5
本实施例提供一种药物中间体的手性提纯方法,所述药物中间体的手性提纯方法包括以下步骤:
(1)在500l反应釜中,将光学纯度为94.77%的式a4所示化合物(55.6kg)加入到甲醇(421kg)中,在10℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为50.5:49.5的混合物),收集滤液;
(2)将步骤(1)得到的滤液在35℃下浓缩后,加入正庚烷(120kg)浓缩,再加入正庚烷(60kg)浓缩,最后再加入正庚烷(60kg)在5℃下混合搅拌0.5h,过滤,滤饼烘干得52.6kg,收率94.6%,得到光学纯度为99.91%的式a4所示化合物。
本实施例提供的光学纯度为94.77%的式a4所示化合物合成路线为:
(1)中间体a1的合成
向2000l反应釜中加入四氢呋喃380kg,cbz-l-丙氨酸96.5kg,hobt70.4kg,二甲羟胺盐酸盐50.3kg,dmap5.3kg,二氯甲烷97kg,10-20℃下搅拌10min,滴加三乙胺110kg,控温20℃以内,滴完后,搅拌30min。加入edc盐酸盐99.3kg,控温20-25℃,保温8h。向体系中加入乙酸乙酯386kg,控温25℃以内,滴加3n盐酸482kg,搅拌后分液,有机相用620kg碳酸氢钠饱和溶液洗,再用150kg饱和氯化钠溶液洗,分液。有机相浓缩干,用500kg正庚烷打浆,抽滤。滤饼烘干得a1103.2kg,收率89.7%,化学纯度99.4%,手性纯度99.8%。
(2)中间体a2的合成
向1000l反应釜中加入a198.8kg,四氢呋喃202kg,3,5-双(三氟甲基)溴苯136.7kg,氮气保护下,降温至-10℃,滴加2m异丙基氯化镁四氢呋喃溶液507kg,控温-5~0℃,滴完后反应2h。在0-10℃下向体系滴加3n盐酸320kg,加入乙酸乙酯278kg,搅拌30min,分层,有机相用130kg饱和食盐水洗,浓缩,再用甲苯60kg*2带2次,得a2粗品181kg黄色油状物。
(3)中间体a3的合成
向a2粗品中加入甲苯300kg,异丙醇160kg,异丙醇铝24.4kg,氮气保护下,升温至50℃,保温16h。把体系降温至10℃以内,加入甲苯600kg,1n盐酸400kg,搅拌30min,分液。有机相浓缩干,加入正庚烷620kg,打浆4h,抽滤。烘干得a3108.6kg,a2、a3两步收率69.5%,化学纯度99.2%,手性纯度94.4%。
(4)光学纯度为94.77%的中间体a4的合成
向2000l反应釜中加入甲苯972kg,异丙醇108kg,a3108kg,降温至10℃以内,加入氢氧化钾28.6kg,升温至20℃,保温2h。体系降温至10℃以内,滴加377kg的1n盐酸,滴完后,搅拌30min,分液。有机相浓缩干,加入432kg正庚烷,控温10-20℃,打浆4h,过滤。滤饼烘干,得a465kg,收率81%,化学纯度99.5%,手性纯度94.77%。
对比例1
本对比例提供一种药物中间体的手性提纯方法,所述光学纯度为94.8%的式a4所示化合物的制备方法同实施例1,所述药物中间体的手性提纯方法包括以下步骤:
(1)在1l的三口烧瓶中,将光学纯度为94.8%的式a4所示化合物(60g)加入到乙醇(150ml)中,在11℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为51.8:48.2的混合物),收集滤液;
(2)将步骤(1)得到的滤液在40℃下浓缩后,烘干得48.4g,收率80.7%,得到光学纯度为97.4%的式a4所示化合物。
对比例2
本对比例提供一种药物中间体的手性提纯方法,所述光学纯度为94.8%的式a4所示化合物的制备方法同实施例1,所述药物中间体的手性提纯方法包括以下步骤:
(1)在1l的三口烧瓶中,将光学纯度为94.8%的式a4所示化合物(66.8g)加入到二氯甲烷(330ml)中,在10℃下混合搅拌0.5h,过滤,滤饼烘干(测得滤饼为a4:a4的质量比为51:49的混合物),收集滤液;
(2)将步骤(1)得到的滤液在35℃下浓缩后,加入正庚烷(200ml)在5℃下混合搅拌0.5h,过滤,滤饼烘干得56.6g,收率84.7%,得到光学纯度为99.0%的式a4所示化合物。
申请人声明,本发明通过上述实施例来说明本发明所述的药物中间体的手性提纯方法,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!