基于噻吩并萘酰亚胺类化合物、噻吩硫醇及其制备与应用
1.本发明涉及有机光电材料领域。更具体地,涉及一种基于噻吩并萘酰亚胺类化合物、噻吩硫醇及其制备与应用。
背景技术:
2.有机半导体材料由于其韧性好,可拉伸,易修饰,应用广泛,易于加工成型、全柔性器件等优点,主要应用于有机场效应晶体管(ofet),太阳能电池(opv),有机发光半导体(oled)等方面。它其中一个最大的优点是可大规模制备,柔性器件,印刷器件,显示了有机材料巨大的发展潜力和广阔的研究前景。
3.有机半导体由于载流子的传输不同分为n型和p型两种。n型半导体的材料由于合成上的困难相对来说发展比较缓慢。萘酰亚胺由于其平面刚性结构,独特的电子离域稳定结构,结构的可调控性,常用于合成n型半导体材料。其一可在n原子上进行烷基链的取代来改变分子的溶解性和固态堆积结构;其二是在溴或硼酯的反应位点进行功能化修饰和分子体系的拓展。
4.在过去的几十年中,供体(d)-受体(a)的材料在有机器件中的应用一直被人们广泛研究。现在人们已开发出不同类型的d
–
a结构,从小分子到共聚物。例如,d
–
a共聚物在太阳能电池中显示出非常高的效率。除此之外,将供体和受体部分连接在一起,提供了一种刚性的平面结构构象,可以减少重组能量,并增强分子间的相互作用,进一步影响分子轨道的分布和晶体堆积,从而得到较高的载流子迁移率。因此,这类分子在有机场效应晶体管(ofet)领域中也可以发挥重要作用。
5.但是,d
–
a-d分子体系在ofet领域合成相对较少。于是本发明设计合成了两个噻吩、三个噻吩甚至更多个噻吩直接稠合在萘酰亚胺母核上,通过增加给体的数目,增加分子的共轭程度,一方面是想通过调节分子排列,增加分子间的作用力,有利与电子传输。另一方面,随着共轭程度的增加,吸收会红移,吸光能力变强,可能会适合做太阳电池材料。
6.基于噻吩直接稠合在萘酰亚胺母核上的有机共轭分子在有机场效应晶体管和太阳能电池中的应用却鲜有报道。有鉴于此,特提出本发明。
技术实现要素:
7.基于以上背景技术,本发明首次设计合成了一种基于噻吩并萘酰亚胺类化合物。萘酰亚胺因具有平面刚性结构,具有电子亲和能高和稳定性强等优点,而广泛应用于有机半导体材料中。其中,噻吩并萘酰亚胺是一类非常重要的n型半导体材料,噻吩的引入可以增加化合物的共振能,拓宽其吸收光谱;噻吩上的活性位点,可以进一步功能化。
8.为了实现以上目的,本发明采用如下技术方案:
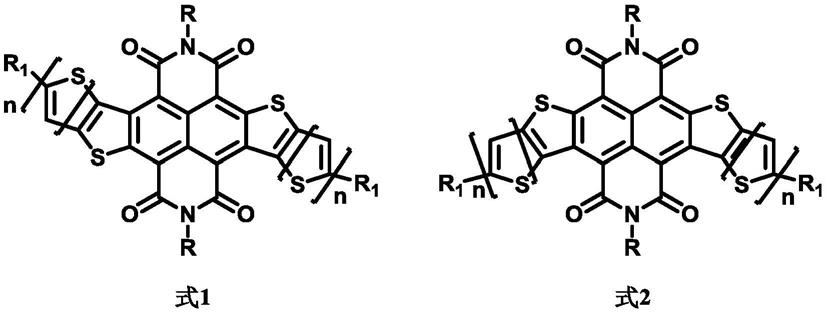
9.所述的基于噻吩并萘酰亚胺类分子化合物的结构式如下所示:
10.本发明第一方面提供一种基于噻吩并萘酰亚胺类化合物,其结构式为式1或式2所示:
[0011][0012]
其中,n为1~5的整数;
[0013]
r基团选自:c
2-60
烷基、含取代基的c
2-60
烷基、c
2-60
烷氧基、含取代基的c
2-60
烷氧基、芳香基、含取代基的芳香基、烷基芳香基、含取代基的烷基芳香基、烷基杂芳香基、含取代基的烷基杂芳香基、烷基杂环基和含取代基的烷基杂环基中的任意一种;
[0014]
r1基团选自:氢原子或硅烷类保护基,例如三异丙基硅基(tips)、三甲基硅基(tms)、三乙基硅基(tes)、叔丁基二甲基硅基(tbdms)、叔丁基二苯基硅基(tbdps)等。
[0015]
优选地,所述取代基选自:甲基、乙基、丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、羟基、巯基、氟原子、氯原子、溴原子、碘原子、氰基、醛基、脂基、磺基、亚磺基、硝基、氨基、亚氨基、羧基和肼基中的至少一种。
[0016]
优选地,所述烷氧基选自:甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、仲丁氧基、异丁氧基、叔丁氧基、戊氧基、己氧基、庚氧基、辛氧基、壬氧基、癸氧基、十一碳烷氧基、十二碳烷氧基、十三碳烷氧基、十四碳烷氧基、十五碳烷氧基、十六碳烷氧基、十七碳烷氧基、十八碳烷氧基、十九碳烷氧基和二十碳烷氧基中的任一种。
[0017]
优选地,所述芳香基选自:苯基、萘基、蒽基、菲基、并四苯基、并五苯基、三苯胺基、芘基、茚基、联苯基和芴基中的至少一种。
[0018]
优选地,所述杂环基、杂芳香基均选自:噻吩、苯并噻吩、吡喃、苯并吡喃、呋喃、苯并呋喃、咪唑、苯并咪唑、吡唑、苯并吡唑、吡咯、苯并吡咯、吡啶、苯并吡啶、吡嗪、苯并吡嗪、吲哚、异吲哚、苯并吲哚、嘧啶、苯并嘧啶、萘啶、苯并萘啶、哒嗪、苯并哒嗪、吲唑、苯并吲唑、嘌呤、苯并嘌呤、喹嗪、苯并喹嗪、喹啉、苯并喹啉、吲嗪、苯并吲嗪、酞嗪、苯并酞嗪、喹喔啉、苯并喹喔啉、噻唑、苯并噻唑、咔啉、苯并咔啉、菲啶、苯并菲啶、菲咯啉、苯并菲咯啉、吖啶、苯并吖啶、吩嗪、苯并吩嗪、吩噻嗪、苯并吩噻嗪、咔唑、苯并咔唑、二并噻吩、二噻吩并吡咯、三并噻吩、四并噻吩和五并噻吩中的任一种。
[0019]
在本发明的一个优选方案中,r选自c
2-60
的烷基、芳香基或含取代基的芳香基,所述烷基选自直链或支链烷基。例如苯基、烷基取代苯基、2,6-双烷基取代的苯基等。
[0020]
优选地,r选自c
2-60
的烷基;或r1选自苯基或含取代基的苯基,所述取代基为c
1-12
的烷基。例如甲基、乙基、正丙基、异丙基、叔丁基、c
10-30
的支链烷基等。
[0021]
更优选地,所述r选自c
1-12
的直链烷基或氟取代的直链烷基、或所述r2、r3选自c
1-12
的烷基,r2、r3相同或不同。
[0022]
更优选地,所述r选自或
[0023]
优选地,r1选自氢原子、三异丙基硅基(tips)或三甲基硅基(tms)。
[0024]
本发明第二方面提供第一方面提供的基于噻吩并萘酰亚胺类化合物的制备方法,该方法的合成路线如下所示:
[0025][0026]
该合成路线中,n为1~5的整数;原料硫醇的结构式表示为具体的,当n为1时,原料硫醇的结构为当n为2时,原料硫醇的结构为当n为3时,原料硫醇的结构为当n为4时,当n为5时,原料硫醇的结构为
[0027]
根据本发明的制备方法,其包括以下步骤:
[0028]
s1、二溴萘酰亚胺与硫醇进行偶联反应,得到中间体1;
[0029]
s2、中间体1在溴化试剂的作用下进行亲核取代溴化,得到中间体2;
[0030]
s3、中间体2进行分子内关环得到产物3和4;此时r1不为氢原子;
[0031]
s4、产物3和4在脱保护基试剂的作用下,脱去三异丙基硅基,得到产物5和6;,r1为氢原子。
[0032]
以下针对每一步进行详细说明:
[0033]
s1、二溴萘酰亚胺与硫醇进行偶联反应,得到中间体1。
[0034]
优选地,s1中,二溴萘酰亚胺与硫醇在n,n-二甲基甲酰胺溶剂中,以碳酸钾作为碱发生一步偶联反应,得到中间体1。
[0035]
s2、中间体1在溴化试剂的作用下进行亲核取代溴化反应,得到中间体2。
[0036]
优选地,s2中所述溴化试剂为溴代琥珀酰亚胺。
[0037]
优选地,s2中所述亲核取代溴化反应的溶剂为三氯甲烷和n,n-二甲基甲酰胺。
[0038]
s3、中间体2进行分子内关环反应得到产物3和4。
[0039]
优选地,s3中所述分子内关环反应在催化剂三(二亚苄基丙酮)二钯,1,8-二氮杂二环十一碳-7-烯作为碱的作用下进行。
[0040]
该步骤产物通过核磁质谱等测试,以及晶体可以证明分子内关环过程中发生了重排,得到了两种分子结构,互为异构体,及产物3和4。
[0041]
s4、产物3和4在脱保护基试剂的作用下,脱去三异丙基硅基,得到产物5和6。
[0042]
优选地,s4中所述脱保护基试剂为四丁基氟化铵。
[0043]
本发明第三方面提供一种噻吩硫醇,其结构式为:
[0044][0045]
其中,n为2~5的整数。
[0046]
优选地,所述噻吩硫醇的结构式为:
[0047][0048]
本发明第四个方面提供第三方面所述噻吩硫醇的制备方法,包括以下步骤:
[0049]
所述噻吩硫醇对应的原料溴取代的噻吩在低温条件下,在丁基锂的作用下拔溴反应,然后加入硫粉取代生成硫醇;
[0050]
所述溴取代的噻吩的取代位点为噻吩硫醇中硫醇所对应的位点。
[0051]
本发明再一方面提供上述基于噻吩并萘酰亚胺类化合物在有机光电器件中的应用。
[0052]
本发明基于噻吩并萘酰亚胺类化合物中,噻吩的引入可以增加化合物的共振能,
拓宽其吸收光谱;噻吩上的活性位点,可以进一步功能化。该基于噻吩并萘酰亚胺类化合物是一种具有重要应用前景的半导体材料。
附图说明
[0053]
图1为本发明合成路线图。
具体实施方式
[0054]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0055]
本发明在所列举出来的优选实施例中所使用的条件均为经过条件优化筛选出的最优条件,并不代表其他所列条件不可实现相应的反应以得到相应的产物,只是产率会有所下降。
[0056]
原料二溴萘酰亚胺的合成路线参考文献j.am.chem.soc.,2012,134(13):5770-5773.
[0057]
实施例1
[0058]
本实施例的合成路线如下所示:
[0059][0060]
原料硫醇1的合成:
[0061]
反应在低温冷凝装置中进行,根据文献路线合成做出3-溴-5-三异丙基硅烷噻吩,将其1g,3.13mmol)入到100ml的两口瓶中,抽换气,加入溶剂无水乙醚50ml。然后放置-78℃的低温冷凝仪里,冷却一段时间后,将丁基锂(2.4m,3.44mmol)溶液逐滴滴加到反应瓶里,
nmr(300mhz,cdcl3))δ163.92,162.00,147.69,140.40,139.32,129.55,129.15,126.60,125.58,124.18,119.29,77.33,77.22,77.02,76.70,44.99,36.47,31.93,31.91,31.64,30.07,29.67,29.60,29.37,29.33,26.46,22.69,18.70,18.46,14.12,11.87,11.60.
[0072]
产物3a和4a的合成:
[0073]
将1,8-二氮杂双环[5.4.0]十一碳-7-烯(0.189g,1.24mmol)加入到100ml两口瓶中,加入中间体2(0.4g,0.311mmol),溶剂为20ml超干n,n-二甲基乙酰胺,抽换气,在氮气保护下加入催化剂三(二亚苄基丙酮)二钯(0.1425g,0.156mmol)催化剂,转移至温度为160℃的油浴锅里。反应加热2小时,体系颜色变为黑色,结束反应后加入三氯甲烷稀释,然后用水将n,n-二甲基乙酰胺溶剂萃取干净,干燥后除去溶剂。二氯甲烷和石油醚体积比为1:1作为淋洗剂,提纯得到纯的化合物紫色,产率为15%。
[0074]
3a的氢谱:1h nmr(400mhz,chloroform-d)δ7.68
–
7.61(m,1h),7.49(d,j=7.8hz,1h),2.86(p,j=6.8hz,2h),1.63
–
1.43(m,12h),1.38
–
1.11(m,21h).
[0075]
4a的氢谱:1h nmr(400mhz,chloroform-d)δ7.71
–
7.57(m,1h),7.52(d,j=7.8hz,1h),2.97
–
2.85(m,2h),2.87
–
2.78(m,3h),1.56(s,12h),1.21(dd,j=40.1,7.1hz,18h).
[0076]
产物3b和4b的合成路线与产物3a和4a相同。
[0077]
3b的氢谱:1h nmr(400mhz,chloroform-d)δ7.79
–
7.62(m,1h),4.41(td,j=23.0,22.0,7.3hz,2h),2.20(s,1h),1.25-1.23(d,j=7.4hz,32h),0.83(dt,j=13.7,6.9hz,6h).
[0078]
4b的氢谱:1h nmr(400mhz,chloroform-d)δ7.67(s,1h),4.40(dd,j=43.7,7.3hz,2h),2.18(s,1h),1.23-1.20(d,j=20.8hz,32h),1.00
–
0.71(m,6h).
[0079]
产物5b和6b的合成:
[0080]
在氮气保护下,将产物3b和4b(0.189g,1.24mmol)溶于四氢呋喃加入到50ml的反应瓶中,加入2ml的四丁基氟化铵(tbaf)溶液,常温搅拌一个小时即可,然后蒸干溶剂,石油醚和甲醇结晶提纯产物,产率为61%。
[0081]
5b的氢谱:1h nmr(400mhz,cdcl3)δ7.88-7.87(d,j=5.3hz,2h),7.44-7.43(d,j=5.29hz,2h),4.58
–
4.21(m,4h),2.32
–
2.09(m,2h),1.56
–
1.13(m,64h),0.99
–
0.74(m,12h).
[0082]
5b的质谱数据hr-maldi-tof(m/z):calcd.for c62h86bn2o4s4:1050.5470,found 1050.54765.
[0083]
实施例2
[0084]
本实施例中n=2,r1=tips。
[0085]
原料硫醇2的合成:
[0086]
由参考文献得到(6-溴噻吩并[3,2-b]噻吩-2)-三异丙基硅烷,将其(2.1g,5.6mmol)加入到100ml的两口瓶中,抽换气,加入溶剂无水乙醚50ml,然后放置-78℃的低温冷凝仪里,冷却搅拌一段时间后,将丁基锂(2.4m,2.5ml)溶液逐滴滴加到反应瓶里,反应一
个小时。然后向溶液中加入升华硫(0.1977g,6.16mmol),在低温下再搅拌两个小时,停掉反应,转移至室温自然升温,在通风处中处理实验,用水淬灭。将所得混合物用(3
×
25ml)naoh 1m萃取。将合并的水层用冰冷却并用10%盐酸酸化以从其钠盐中释放出硫醇。用乙醚(3
×
50ml)萃取产物。加入无水硫酸钠干燥,蒸干溶剂,得到黄色固体。
[0087]
中间体1c的合成
[0088][0089]
反应在100℃油浴锅中进行,准备一个100ml的两口瓶,将上一步做好得硫醇2(1.5g,3.9mmol)滴加到反应瓶中,注意硫醇有难闻的气味。然后加入二溴异丙基苯链的ndi(0.7g,9.41mmol)和碳酸钾(0.65g,4.7mmol),选用超干的n,n-二甲基甲酰胺20ml作为反应溶剂。抽换气,在氮气保护下反应,将反应瓶转移到100℃的油浴锅里反应,可以看到一会溶液的颜色变深,变红。反应两个半小时后,停掉反应,冷却至室温。将反应液用三氯甲烷稀释,然后用水萃取,用水洗三遍,再用饱和食盐水洗。然后用无水硫酸钠干燥过滤后,用旋转蒸发仪旋干溶剂,过柱子提纯粗产物,淋洗剂为二氯甲烷和石油醚2:1,蒸干溶剂得到纯的化合物红色固体,产率为60%。
[0090]1h nmr(400mhz,chloroform-d)δ8.23(s,1h),7.83(s,1h),7.47
–
7.39(m,2h),7.29(d,j=7.8hz,2h),2.62(s,2h),1.29(dd,j=14.8,7.3hz,3h),1.24
–
0.96(m,30h).
[0091]
中间体2c的合成
[0092][0093]
反应在60℃进行,将上一步得到的中间体1c(0.46g,0.37mmol),溶解在三氯甲烷和n,n-二甲基甲酰胺(1:1)溶剂中,抽换气,在氮气保护下,油浴锅调到温度60℃。分批加入nbs(0.26g,1.46mmol)上溴,反应过程中不断点板,观察反应进程,可以很明显看到原料上方有新点生成,当没有原料剩余时,停掉反应。反应液冷至室温后,倒入水中用氯仿萃取,洗
三次。然后用二氯甲烷和石油醚作为淋洗剂提纯,得到红色产物,产率为(60%)。
[0094]1h nmr(400mhz,chloroform-d)δ8.17(s,1h),7.46(t,j=7.8hz,1h),7.37
–
7.28(m,2h),2.68
–
2.61(m,2h),1.29(dt,j=13.0,6.6hz,3h),1.18(d,j=6.8hz,12h),1.08(t,j=6.2hz,18h).
[0095]
产物3c和4c的合成
[0096][0097]
反应温度为160℃的高温反应,准备一个100ml的两口瓶,将上一步得到的中间体2c(0.3g,0.21mmol)和碱1,8-二氮杂双环[5.4.0]十一碳-7-烯(0.13g,0.85mmol)放进反应瓶中。加入溶剂超干n,n-二甲基乙酰胺溶剂(25ml),抽换气,在氮气氛围中加入催化剂三(二亚苄基丙酮)二钯(0.09g,0.098mmol),放在温度为160℃的油浴锅里。混合物加热两个小时,反应变黑色,停掉反应后,冷却至室温,加入三氯甲烷充分溶解,倒入水中萃取三次,饱和食盐水萃取三次。用无水硫酸钠干燥,没有水分后过滤后旋干溶剂。得到蓝色粗产物,然后过柱子提纯,淋洗剂为二氯甲烷和石油醚2:1,分离得到纯的蓝色化合物,产率为10%。
[0098]
产物5c和6c的合成
[0099][0100]
在氮气保护下,将产物3c和4c(0.189g,1.24mmol)溶于四氢呋喃加入到50ml的反应瓶中,加入2ml的四丁基氟化铵(tbaf)溶液,常温搅拌一个小时即可,然后蒸干溶剂,石油醚和甲醇结晶提纯产物,产率为61%。
[0101]
实施例3
[0102]
n=3,r1=tips。
[0103]
原料硫醇3的合成:
[0104]
根据文献报道合成(5-溴二噻吩并[3,2-b]噻吩-2)-三异丙基硅烷i(1g,2.32mmol)到100ml的两口瓶中,抽换气,在氮气保护下取用提前重蒸的乙醚50ml,然后放置-78℃的低温冷凝仪里,冷却一段时间后,将丁基锂(2.4m,1.02ml)溶液逐滴滴加到反应瓶里,反应两个小时。然后加入升华硫(0.082g,2.56mmol)在低温下再搅拌两个小时,直至颜色澄清,为黄色溶液。停掉反应,转移至室温自然升温,然后用水淬灭。在水层用10%盐酸酸化释放出硫醇。用乙醚(3
×
50ml)萃取产物,有机层加入无水硫酸钠干燥,蒸干溶剂,得到红色固体。
[0105]
中间体1d的合成:
[0106][0107]
反应在油浴锅中进行,准备一个100ml的两口瓶,将上一步做好得硫醇3(0.54g,1.41mmol)滴加到反应瓶中,然后加入二溴ndi(0.35g,0.47mmol)和碳酸钾,溶剂为超干的n,n-二甲基甲酰胺30ml。抽换气,在氮气氛围下反应,将反应瓶转移到100℃的油浴锅里反应,可以看到一会溶液的颜色变深,变红色。反应两个小时后,停掉反应,冷却至室温。将反应液倒入水中,用二氯甲烷萃取三次,再用饱和食盐水洗三次,直至全部萃取干净。无水硫酸钠干燥过滤后,旋干溶剂,过柱子提纯粗产物,淋洗剂为二氯甲烷和石油醚2:1,蒸干溶剂得到纯的化合物红色固体,产率为50%。
[0108]1h nmr(400mhz,chloroform-d)δ8.49(s,1h),7.69(s,1h),7.42(d,j=11.0hz,2h),7.34
–
7.24(m,2h),2.63(p,j=6.8hz,2h),1.48
–
0.99(m,3h),0.87(dtd,j=12.2,6.2,5.6,2.8hz,30h).
[0109]
中间体2d的合成:
[0110][0111]
将上一步得到的1d(0.23g,0.17mmol),溶解在三氯甲烷中,充分溶解后加入n,n-二甲基甲酰胺(2:1)溶剂中,抽换气,再氮气保护下,温度为70℃。分批加入nbs(0.12g,0.67mmol)上溴,反应过程中不断点板,观察反应进程,当没有原料剩余时,上方有上两个溴的点,停掉反应。反应液冷至室温后,倒入水中氯仿萃取,洗三次。然后用二氯甲烷和石油醚作为淋洗剂提纯,得到红色产物,产率为(60%)。
[0112]1h nmr(600mhz,chloroform-d)δ8.53(s,1h),7.66(s,1h),7.44(t,j=7.7hz,2h),7.30(d,j=7.8hz,2h),2.63(q,j=6.8hz,2h),1.84(p,j=7.5hz,3h),1.30
–
1.05(m,30h).
[0113]
产物3d和4d的合成
[0114][0115]
选用100ml的两口瓶,将1,8-二氮杂双环[5.4.0]十一碳-7-烯(0.02g,0.13mmol)加入到化合物2d(0.05g,0.03mmol)中,加入超干n,n-二甲基乙酰胺溶剂(10ml),在氮气氛围下,加入(二亚苄基丙酮)二钯(0.015g,0.016mmol)催化剂,放在温度为160℃的油浴锅里。混合物加热一个小时,反应很快,溶液变为黑色,停掉反应后,冷却至室温。加入三氯甲烷充分溶解,倒入水中萃取三次,饱和食盐水萃取三次。用无水硫酸钠干燥后旋干溶剂。溶解度还可以,硅胶柱提纯,淋洗剂为三氯甲烷和石油醚体积比1:1,提纯化合物,得到黑色固体,产率为7%。
[0116]
产物5d和6d的合成
[0117][0118]
合成步骤与5c和6c相同。
[0119]
实施例4
[0120]
本实施例中n=1,r=-c8h
17
,r1=tips。
[0121]
中间体1f的合成:
[0122][0123]
合成步骤同1a。
[0124]1h nmr(400mhz,chloroform-d)δ8.10(s,1h),8.00(d,j=0.8hz,1h),7.24(d,j=0.9hz,1h),4.18
–
4.08(m,2h),1.75
–
1.60(m,3h),1.36(dd,j=14.9,7.3hz,12h),1.14(d,j=7.4hz,18h),0.86(s,3h).
[0125]
中间体2f的合成:
[0126][0127]
合成步骤同2a。
[0128]1h nmr(400mhz,chloroform-d)δ8.03(s,1h),7.17(s,1h),4.27
–
4.04(m,2h),1.71(s,2h),1.50
–
1.20(m,3h),1.13(d,j=7.3hz,30h),0.88(t,j=7.7hz,3h).
[0129]
产物3f和4f的合成:
[0130][0131]
合成路线同3a和4a。
[0132]
3f的氢谱:1h nmr(400mhz,chloroform-d)δ7.69(s,1h),4.52
–
4.32(m,2h),1.90(q,j=7.8hz,3h),1.69
–
1.16(m,30h),0.97
–
0.79(m,3h).
[0133]
4f的氢谱:1h nmr(400mhz,chloroform-d)δ7.69(s,1h),4.46(dt,j=43.8,7.6hz,2h),1.92(d,j=8.6hz,3h),1.66
–
1.15(m,30h),0.88(dq,j=7.0,4.0,3.0hz,3h).
[0134]
产物5f和6f的合成路线与5a和6a一样。
[0135]
5f的质谱数据hr-maldi-tof(m/z):calcd.for c38h38bn2o4s4:714.1714,found 714.17220.
[0136]
6f的质谱数据hr-maldi-tof(m/z):calcd.for c38h38bn2o4s4:714.1714,found 714.17202.
[0137]
实施例5
[0138]
本实施例中n=1,r1=tms。
[0139]
合成路线如下所示,具体过程如以上实施例。
[0140][0141]
实施例6
[0142]
本实施例中n=1,r1=tips。
[0143]
中间体1h的合成:
[0144][0145]
合成路线与1a相同。
[0146]
1h nmr(400mhz,chloroform-d)δ8.15(s,1h),8.05(s,1h),4.99(t,j=15.3hz,2h),1.57(s,3h),1.47
–
0.80(m,18h).
[0147]
中间体2h的合成:
[0148][0149]
合成路线与2a相同。1h nmr(400mhz,chloroform-d)δ8.09(s,1h),7.20(s,1h),5.01(t,j=15.3hz,2h),1.48
–
0.74(m,21h).
[0150]
产物3h和4h的合成:
[0151][0152]
合成路线与3a相同。3h的氢谱:1h nmr(400mhz,chloroform-d)δ7.69(s,1h),5.31(t,j=15.4hz,2h),1.55-1.53(s,3h),1.24-1.22(m,18h).
[0153]
4h的氢谱:1h nmr(400mhz,chloroform-d)δ7.68(s,1h),5.34(t,j=15.2hz,1h),5.22(d,j=15.2hz,1h),1.58-1.54(m,3h),1.25-1.23(m,18h).
[0154]
产物5h和6h的合成:
[0155][0156]
合成路线与5a和6a相同。
[0157]
5h的质谱数据hr-maldi-tof(m/z):calcd.for c30h8f14n2o4s4:853.914884,found 853.91374.
[0158]
6h的质谱数据hr-maldi-tof(m/z):calcd.for c30h8f14n2o4s4:853.914884,found 853.91468.
[0159]
以上实施例中的硫醇的起始原料按照以下文献合成:zhang x,adrien p,matzger a j.synthesis and structure of fusedα-oligothiophenes with up to seven rings[j].journal of the american chemical society,2005,127(30):10502-10503.
[0160]
本领域技术人员可以理解的,本发明内容部分所列出的取代基的胺,均为实验中常用的胺试剂。而本发明中的r1可以选择发明内容中的其他不同取代基,其对应的二溴萘酰亚胺的合成,参照文献j.am.chem.soc.,2012,134(13):5770-5773等均可实现。
[0161]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发
明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1