西那卡塞中间体及其合成方法、应用与流程
本发明涉及医药技术领域,具体涉及一种西那卡塞中间体及其合成方法、应用。
背景技术:
继发性甲状旁腺功能亢进症(shpt)是慢性肾功能衰竭患者常见的并发症,也是慢性肾衰竭终末期血液透析时最主要、最严重的并发症之一。其主要表现为甲状旁腺激素(pth)水平升高和甲状旁腺增生,可导致严重的骨骼损害、难治性皮肤瘙痒、贫血、神经系统损害及心血管疾病等。研究显示,pth的长期高水平状态可增加慢性肾病需要长期透析患者的死亡风险。近年来对shpt的发病机制在分子水平上有了新的认识,认为甲状旁腺钙敏感受体等发挥着重要的作用。shpt以矿物质代谢紊乱、甲状旁腺激素分泌增多、甲状旁腺增生为特征,它可引起一系列后果,包括肾性骨营养不良、血管和心脏瓣膜钙化等。
盐酸西那卡塞(cinacalcethydrochloride),由美国npspharmaceuticals公司研发的拟钙剂,化学名称为n-[(1r)-1-(1-萘基)乙基]-3-[3-(三氟甲基)苯基]丙-1-胺,2004年fda批准amgen公司生产的西那卡塞盐酸盐上市,用于治疗肾病透析患者的继发性甲状旁腺机能亢进和甲状腺癌所致的高钙血症。该药能激活甲状旁腺中的钙受体,从而降低甲状旁腺素(pth)的分泌,进而导致血清钙及磷酸钙产物水平的降低。该药物结构式如下:
本发明所涉及的一种(r)-1-(1-萘基)乙胺是盐酸西那卡塞的中间体之一,目前制备(r)-1-(1-萘基)乙胺的制备方法有化学法和酶法:
专利(cn101735070a)中报道了一种(r)-1-(1-萘基)乙胺的拆分方法。该方法反应比较繁琐,过程长,合成过程中采用多次拆分以及消旋的操作,导致反应过程中原料损耗较大。
也有一些酶法拆分,如以下反应式所示(r)-1-(1-萘基)乙胺在脂肪酶作用下酯化,同时(s)-1-(1-萘基)乙胺在钯催化剂作用下加氢消旋,拆分和消旋在-一个步骤中进行。(r)-1-(1-萘基)乙胺酯化物水解萃取后得到(r)-1-(1-萘基)乙胺。虽然该工艺路线较短,但所使用的消旋催化剂是一些钯催化剂等贵金属的复合物,这些消旋催化剂成本极高,且拆分制备过程中使用这些消旋催化剂时,同时钯催化剂容易脱落,重复利用性差;同时存在一些金属钯与酶直接接触,导致酶易于失活,提高了成本的问题。
技术实现要素:
本发明的目的是提供一种西那卡塞中间体及其合成方法、应用。
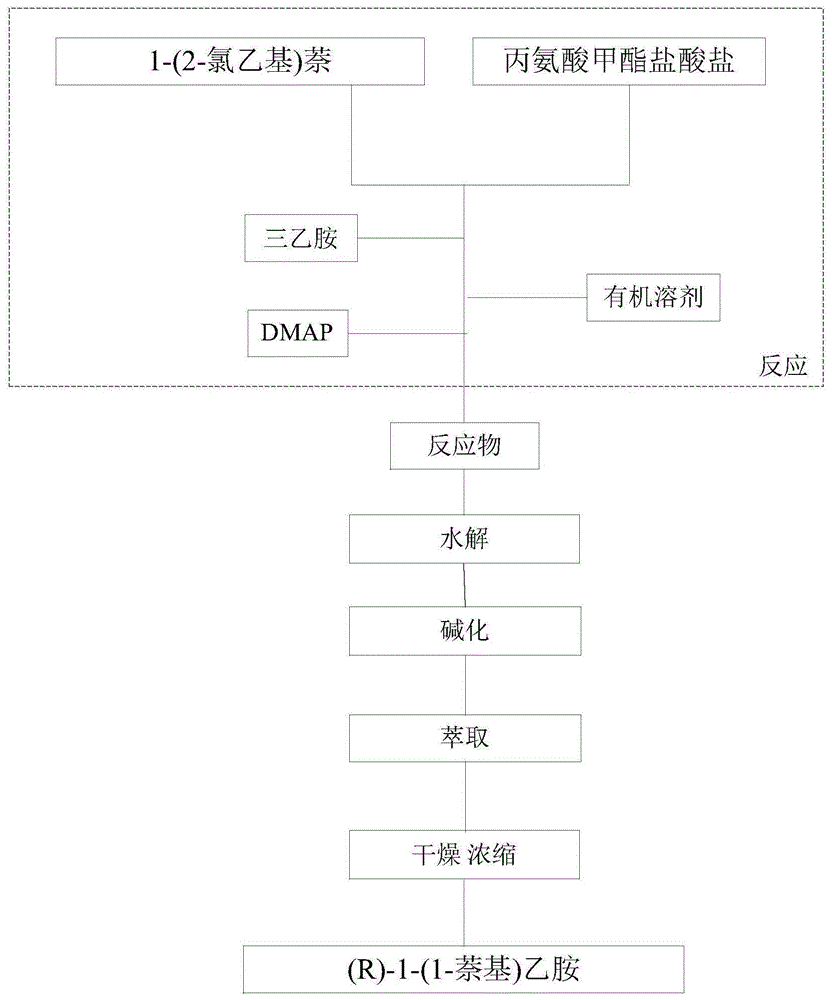
为了解决上述技术问题,本发明提供了一种(r)-1-(1-萘基)乙胺的合成方法,包括:通过1-(2-氯乙基)萘、丙氨酸甲酯盐酸盐制备r-2-萘丙氨酸甲酯;以及对r-2-萘丙氨酸甲酯进行水解、碱化、萃取、浓缩,得到所述(r)-1-(1-萘基)乙胺。
进一步,所述制备r-2-萘丙氨酸甲酯包括:将1-(2-氯乙基)萘与丙氨酸甲酯盐酸盐以三乙胺作为缚酸剂、dmap作为催化剂,在有机溶剂中20-30℃反应,反应结束后过滤、浓缩得到反应物r-2-萘丙氨酸甲酯,待用。
进一步,反应的反应式为:
进一步,1-(2-氯乙基)萘与丙氨酸甲酯盐酸盐的摩尔比值为1:1.1-1.2。
进一步,1-(2-氯乙基)萘与三乙胺的摩尔比值为1:1.05-1.15。
进一步,1-(2-氯乙基)萘与催化剂dmap的质量比值为1:1.5%-2%。
进一步,所述有机溶剂为甲醇或者二氯甲烷。
进一步,所述水解包括:将r-2-萘丙氨酸甲酯在4-4.5倍的4mol/l的hcl中95-105℃回流,水解。
进一步,水解的反应式为:
又一方面,本发明还提供了一种(r)-1-(1-萘基)乙胺,所述(r)-1-(1-萘基)乙胺的结构式为:
第三方面,本发明还提供了一种(r)-1-(1-萘基)乙胺作为合成西那卡塞中间体的应用。
本发明的有益效果是,本发明的西那卡塞中间体及其合成方法、应用通过选用1-(2-氯乙基)萘为初始原料、丙氨酸甲酯盐酸盐为手性源,结合后续添加的反应物进行反应,最终合成(r)-1-(1-萘基)乙胺,通过控制反应的反应条件(如反应条件、反应物加入时间、反应温度等)可以有效提高反应速率,缩短反应时间;通过合理设置各原料的组分含量配比,可以有效提高产物纯度和收率。本合成方法合成路线短,综合成本低,环境压力小,且最终产物纯度高达99%,收率可达89%以上,有利于工业化生产。
附图说明
下面结合附图和实施例对本发明进一步说明。
图1是本发明(r)-1-(1-萘基)乙胺的合成方法的工艺流程图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
如图1所示,本实施例1提供了一种(r)-1-(1-萘基)乙胺的合成方法,包括:步骤s1,通过1-(2-氯乙基)萘、丙氨酸甲酯盐酸盐制备r-2-萘丙氨酸甲酯;以及步骤s2,对r-2-萘丙氨酸甲酯进行水解、碱化、萃取、浓缩,得到所述(r)-1-(1-萘基)乙胺。
具体的,本实施例1通过选用1-(2-氯乙基)萘为初始原料、丙氨酸甲酯盐酸盐为手性源,结合后续添加的反应物进行反应,最终合成(r)-1-(1-萘基)乙胺,通过合理设置各原料的组分含量配比,可以有效提高产物纯度和收率;最终产物纯度高达99%,收率可达89%以上,有利于工业化生产。
作为反应的一种可选的实施方式。
所述制备r-2-萘丙氨酸甲酯包括:将1-(2-氯乙基)萘与丙氨酸甲酯盐酸盐以三乙胺作为缚酸剂、dmap作为催化剂,在有机溶剂中20-30℃反应,反应结束后过滤、浓缩得到反应物r-2-萘丙氨酸甲酯,待用。
进一步,反应的反应式为:
进一步,1-(2-氯乙基)萘与丙氨酸甲酯盐酸盐的摩尔比值为1:1.1-1.2。
进一步,1-(2-氯乙基)萘与三乙胺的摩尔比值为1:1.05-1.15。
进一步,1-(2-氯乙基)萘与催化剂dmap的质量比值为1:1.5%-2%。
进一步,所述有机溶剂为甲醇或者二氯甲烷。
进一步,所述水解包括:将r-2-萘丙氨酸甲酯在4-4.5倍的4mol/l的hcl中95-105℃回流,水解。
进一步,水解的反应式为:
实施例2
在实施例1的基础上,本实施例2提供了一种(r)-1-(1-萘基)乙胺,所述(r)-1-(1-萘基)乙胺的结构式为:
关于(r)-1-(1-萘基)乙胺的组分含量及具体实施过程参见实施例1的相关论述,此处不再赘述。
实施例3
在实施例1的基础上,本实施例3提供了一种(r)-1-(1-萘基)乙胺作为合成西那卡塞中间体的应用。
关于(r)-1-(1-萘基)乙胺的组分含量及具体实施过程参见实施例1的相关论述,此处不再赘述。
实施例4
本实施例4列举了三组实验,对三组实验合成的产物(r)-1-(1-萘基)乙胺的纯度和收率的影响因素进行了探究,如表1所示。
表1组分含量及产物收率
组1
(1)将100g的1-(2-氯乙基)萘溶于500ml甲醇中,并依次加入55.7g三乙胺,80.5g丙氨酸甲酯盐酸盐,以及1.5g的dmap作催化剂,温度20-30℃,反应8小时,tlc检测,反应结束后,过滤掉固体,滤液浓缩至干,得到112.5gr-2-萘丙氨酸甲酯。
(2)将所得112.5gr-2-萘丙氨酸甲酯加入到450ml的4mol/l盐酸中,升温到95℃回流,保持温度反应8小时,反应结束后,降温到20-25℃,水层用饱和naoh溶液水调节ph值>10后用乙酸乙酯萃取2次,每次200ml,萃取结束将酯层用无水硫酸钠干燥30分钟,浓缩后得到80.2g(r)-1-(1-萘基)乙胺,纯度为99.249%,收率为89.5%。
组2
(1)将100g的1-(2-氯乙基)萘溶于500ml甲醇中,并依次加入58.4g三乙胺,84.2g丙氨酸甲酯盐酸盐,以及1.75g的dmap作催化剂,温度20-30℃,反应8小时,tlc检测,反应结束后,过滤掉固体,滤液浓缩至干,得到114.6gr-2-萘丙氨酸甲酯。
(2)将所得114.6gr-2-萘丙氨酸甲酯加入到460ml的4mol/l盐酸中,升温到95℃回流,保持温度反应8小时,反应结束后,降温到20-25℃,水层用饱和naoh溶液水调节ph值>10后用乙酸乙酯萃取2次,每次200ml,萃取结束将酯层用无水硫酸钠干燥30分钟,浓缩后得到(r)-1-(1-萘基)乙胺,纯度为99.75%,收率为91.7%。
组3
(1)将100g的1-(2-氯乙基)萘溶于500ml甲醇中,并依次加入61g三乙胺87.9g丙氨酸甲酯盐酸盐,以及2g的dmap作催化剂,温度20-30℃,反应8小时,tlc检测,反应结束后,过滤掉固体,滤液浓缩至干,得到115.8gr-2-萘丙氨酸甲酯。
(2)将所得115.8gr-2-萘丙氨酸甲酯加入到460ml的4mol/l盐酸中,升温到95℃回流,保持温度反应8小时,反应结束后,降温到20-25℃,水层用饱和naoh溶液水调节ph值>10后用乙酸乙酯萃取2次,每次200ml,萃取结束将酯层用无水硫酸钠干燥30分钟,浓缩后得到(r)-1-(1-萘基)乙胺,纯度为99.75%,收率为92.8%。
综上所述,本发明的西那卡塞中间体及其合成方法、应用通过选用1-(2-氯乙基)萘为初始原料、丙氨酸甲酯盐酸盐为手性源,结合后续添加的反应物进行反应,最终合成(r)-1-(1-萘基)乙胺,通过控制反应的反应条件(如反应条件、反应物加入时间、反应温度等)可以有效提高反应速率,缩短反应时间;通过合理设置各原料的组分含量配比,可以有效提高产物纯度和收率。本合成方法合成路线短,综合成本低,环境压力小,且最终产物纯度高达99%,收率可达89%以上,有利于工业化生产。
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种丙辛吲哚盐酸盐药物中间体丙辛吲哚的合成方法与制造工艺
- 一种吲哚洛尔药物中间体(一)‑4‑(2‑羟基‑3‑异丙氨基丙氧)吲哚的合成方法与制造工艺
- 依替米贝的中间体及其合成方法与依替米贝的合成方法与制造工艺
- 一种白消安药物中间体1,4‑双甲基磺酸丁二酯的合成方法与制造工艺
- 一种塞利洛尔药物中间体3‑(4‑乙氧基苯基)‑1,1‑二乙基脲的合成方法与制造工艺
- 一种羟基保泰松药物中间体4‑羟基偶氮苯的合成方法与制造工艺
- 一种甲氧卡那霉素药物中间体γ‑氨基‑α‑羟基丁酸的合成方法与制造工艺
- 一种非那西丁药物中间体对硝基苯乙醚的合成方法与制造工艺
- 一种间尼索地平药物中间体乙酰乙酸异丁酯的合成方法与制造工艺
- 一种甲氧苄啶药物中间体3,4,5—三甲氧基苯甲酸甲酯的合成方法与制造工艺