基于CL7-CVN和Im7系统分离、富集和检测囊膜病毒的方法与流程
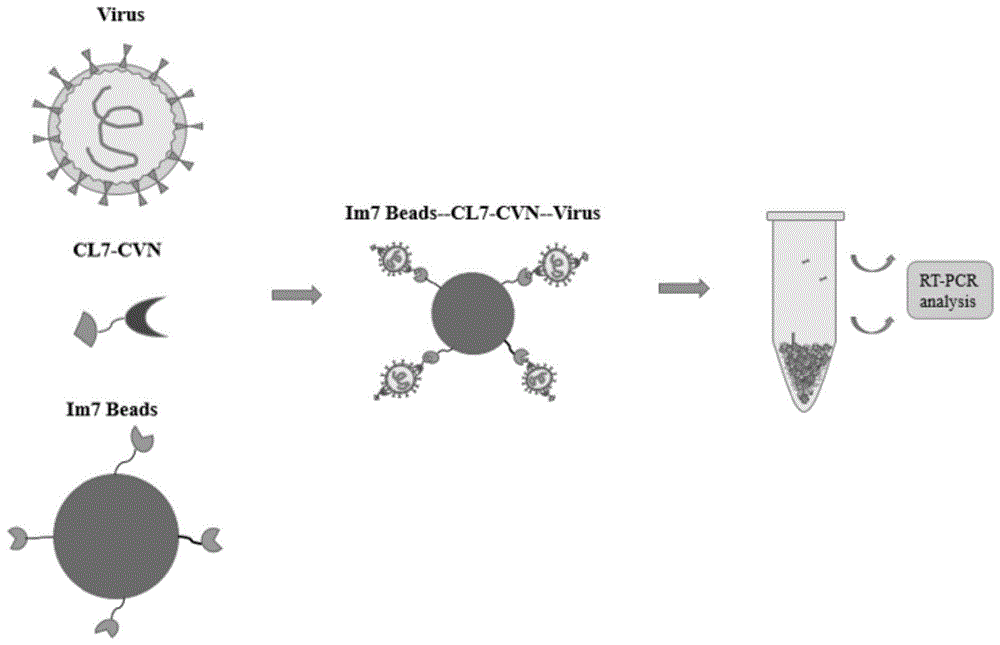
本发明属于生物技术应用领域和环境监测技术领域,具体涉及一种基于cl7-cvn和im7系统分离、富集和检测囊膜病毒的方法。
背景技术:
目前,如艾滋病病毒、单纯疱疹病毒、丙型肝炎病毒、流感病毒、伪狂犬病毒、非洲猪瘟、埃博拉病毒等囊膜病毒,已经给人类生存和发展制造了巨大的威胁,甚至灾难。现有囊膜病毒的分离诊断方法有很多,如病毒分离培养、pcr核酸检测、酶联免疫吸附测定等;然而,这些方法均存在操作复杂、费时,对技术要求高且价格昂贵,不利于快速和现场检测等缺点。因此建立一个简便快速、准确、高通量的诊断检测方法,对控制病毒暴发流行具有重要意义。
技术实现要素:
cyanovirin-n(简称cvn)是一种从蓝藻中分离得到的一种水溶性糖蛋白,能特异地、高亲和性地结合在囊膜病毒衣壳蛋白上阻止病毒感染入侵细胞,从而发挥抗病毒活性,这一特点使它可以不受病毒变异的干扰,同时它还具有抗病毒谱广、性质稳定等特点,这使得cvn蛋白成为一种很有价值的抗病毒药物,同时也为其成为分离检测囊膜病毒的方法成为可能。研究表明,cl7/im7是目前发现的最强蛋白相互作用的1组蛋白(kd为10-14-10-17m)。
本发明通过基因工程方法将cl7融合至cvn蛋白上,得到融合cl7标签的重组蛋白cl7-cvn。它既能够显著提高cvn可溶性表达水平,并且表达的cl7-cvn融合蛋白具有显著的抗病毒活性,而且重组cl7-cvn蛋白还能特异性地结合im7蛋白。
根据通过cl7/im7超高亲和系统,重组cl7-cvn能够同时特异地、高亲和性地结合囊膜病毒衣壳蛋白及和im7。进一步地,将im7结合至微球颗粒上,能够将溶液中病毒颗粒特异性地结合至微球颗粒表面,从而达到从溶液中分离病毒颗粒的效果,并以此建立了一个分离富集检测囊膜病毒检测方法。此方法具有灵敏度高、分离速度快、特异性强、操作简单、可重复性好,不需要昂贵的仪器设备等优点。
本发明提供的一种富集、分离和检测囊膜病毒的方法,其技术方案如下:
一种基于cl7-cvn和im7系统分离和富集囊膜病毒的方法,包括以下步骤:
(1)将cl7-cvn的表达载体转化至第一宿主细胞中,诱导表达,纯化后得到重组cl7-cvn蛋白;
(2)将im7的表达载体转化至第二宿主细胞中,诱导表达,纯化后得到im7蛋白;
(3)将im7蛋白与微球偶联反应,得到im7-beads;
(4)先向待分离富集样品中投入重组cl7-cvn蛋白,孵育后,再投入im7-beads,孵育后富集,离心得到富集病毒的im7-beads。
一种检测囊膜病毒的方法,提取所述的im7-beads富集的囊膜病毒核酸,采用荧光定量pcr检测富集的病毒拷贝数,并计算囊膜病毒的富集效率。
本发明提供的技术方案至少包括以下有益效果:
1、本发明通过基因工程的方法制备了im7蛋白,并以此制备了im7-beads微球,im7-beads微球平均粒径为82.35±0.35μm至151.27±0.48μm之间,能够提供更优异的微观结构和溶胀性能,为其富集和分离病毒提供更佳的效果。
2、本发明通过基因工程的方法制备了重组cl7-cvn蛋白,并利用cl7与im7的特异性结合、及cvn结合囊膜病毒的性能,能够将prv病毒颗粒富集至im7-beads微球表面,为囊膜病毒的分离、富集和检测提供一种全新的技术手段;并且im7-beads微球富集病毒的容量达到1.06×1010viruses/ml。
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
附图说明
图1为本发明提供的富集和分离病毒的原理图;
图2为本发明提供的线性载体和目的基因片段pcr扩增结果电泳图(sds-page):图中1、2泳道分别为原始载体pet28a、pcr扩增得到的线性载体pet28a;3、4泳道分别为pcr扩增得到cl7-linker-his、3c-cvn线性片段,m泳道为dl5000marker;
图3为本发明提供的转化后大肠杆菌dh5a菌落的pcr鉴定结果电泳图(sds-page):1泳道为原始载体pcr对照;2、3、4、5泳道分别为不同菌落的pcr鉴定结果;m泳道为dl5000marker;
图4为本发明提供的转化后质粒鉴定结果电泳图(sds-page):1泳道为pcr扩增得到的线性pet28a载体;2、3、4、5泳道分别为不同菌落的质粒鉴定结果;m泳道为dl5000marker;
图5为本发明实施例提供的cl7-cvn蛋白纯化结果:1泳道,热处理后的上清蛋白;2泳道,挂柱后流穿;3泳道,洗脱的cl7-cvn;m蛋白marker;
图6为本发明实施例提供的im7蛋白的表达纯化结果:wcl泳道,pet28a-im7表达的全蛋白;sup泳道,细胞裂解后的上清蛋白;pel泳道,细胞裂解后沉淀蛋白;h-sup泳道,细胞裂解上清蛋白热处理离心后的上清蛋白;el1,el2,el3,洗脱的im7蛋白;m蛋白marker;
图7为本发明实施例提供的elisas检测cl7、cvn、cl7-cvn与病毒的结合结果:图7a,elisas验证cl7-cvn与prv结合的原理图;图7b,等摩尔的cl7、cvn、cl7-cvn稀释不同倍数后与病毒结合后,用anti-hishrp抗体检测结合的cl7-cvn、cvn,测得的od450值;
图8为本发明实施例提供的cl7-cvn与im7-beads微球富集prv荧光检测:ep管1为仅有im7-beads微球的空白对照;ep管2中仅有prv病毒液;ep管3中为im7-beads微球与prv病毒液混合状态;ep管4为ep管3洗涤im7-beads微球后的状态;ep管5为im7-beads微球、prv病毒液与cl7蛋白液混合状态;ep管6为ep管5洗涤im7-beads微球后的状态;ep管7为im7-beads微球、prv病毒液与cvn蛋白液混合状态;ep管8为ep管7洗涤im7-beads微球后的状态;ep管9为im7-beads微球、prv病毒液与cl7-cvn蛋白液混合状态;ep管10为ep管9洗涤im7-beads微球后的状态;图8中上排为白光照射图,下排为蓝光照射下荧光图;
图9为本发明实施例提供的rt-pcr检测病毒富集效率结果;等摩尔的cl7、cvn、cl7-cvn与等量的im7微球,富集prv的效率;
图10为本发明实施例提供的不同实施例制备的微球吸附病毒的富集效率结果;
图11为本发明实施例提供的pcr检测富集到的prv基因片段:图11a,gd基因的片段(226bp),图11b,gb基因片段(331bp);1泳道,阳性对照;2泳道,水对照;3泳道,pbs与im7-beads处理组;4泳道,cl7与im7-beads处理组;5泳道,cvn与im7-beads处理组;6泳道,cl7-cvn与im7-beads处理组;
图12为本发明实施例提供的病毒富集和分离条件的优化;不同质量的cl7-cvn处理病毒对im7-beads富集分离病毒的影响:0,0.125,0.25,0.5,1μmolcl7-cvn处理分离病毒rt-pcr检测病毒富集效率结果;
图13为本发明实施例提供的病毒液样本经过富集分离后上清病毒感染性检测:正常细胞;病毒原液感染;im7-beads非特异性吸附;im7-beads与cl7-cvn的吸附;
图14为本发明实施例提供的富集和分离病毒的敏感性检测结果;图14a,rt-pcr检测不同稀释体积病毒样本的病毒富集效率;图14b,pcr检测不同稀释体积病毒样本富集后病毒的gk基因片段检测;
图15为本发明实施例提供的富集和分离病毒的容量测定结果;图15a,病毒样本体积从100μl增加到500μl时,病毒的富集量;图15b,病毒样本体积从100μl增加到500μl时,病毒的富集效率。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明作进一步地详细描述。显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,均属于本发明保护的范围。
本发明实施例涉及的主要材料如下:
大肠杆菌rosetta(de3)(merck公司)、大肠杆菌bl21(de3)(merck公司)、pet28a质粒(merck公司)、pet23a质粒(merck公司)。3c蛋白酶、pfudna聚合酶由本实验室表达纯化,t5核酸外切酶购自neb公司,超低分子蛋白质分子量标准蛋白购自博士德公司、引物购自上海生工生物公司。伪狂犬病毒(pseudorabiesvirus,prv)、pk15细胞由华中农业大学赠予本实验室保存。pbs缓冲液(8mmna2hpo4,136mmnacl,2mmkh2po4,2.6mmkcl),咪唑缓冲液(5-200mmimidazole,40mmnah2po4,300mmnacl),1miptg溶液,3c酶切缓冲液(25mmtris-hcl,ph7.0,150mmnacl,0.5mmedta,and1mmdtt)。
重组cl7-cvn蛋白原核表达载体的构建
1、第一pcr扩增:通过定点突变得到突变体cl7基因(核苷酸序列如seqidno:3所示,突变体蛋白氨基酸序列如seqidno:4所示);
以pet23a-cl7质粒为模板,如seqidno:6所示的f1和如seqidno:7所示的r1为引物同时引入linker、his及末端同源序列,进行pcr扩增,得到cl7-linker-his线性片段,参见图2中3泳道;根据genebank公布的cvn基因序列,进行密码子优化设计合成的cvn基因为模板,以如seqidno:8所示的f2和如seqidno:9所示的r2为引物,同时引入3c及末端同源序列,进行pcr扩增,得到3c-cvn线性片段,参见图2中4泳道。引物设计如下:
f1:5’-agaaggagatataccatgggcagcaaaagca-3’,如seqidno:6所示;
r1:5’gtgatggtgatgactgccccctccaccgctccctccgccaccgccttcaatatcaatgtt-3’,如seqidno:7所示;
f2:5’agtcatcaccatcaccatcacttggaggttttgttccagggtccacttggtaaattctccc-3’,如seqidno:8所示;
r2:5’-cggatcctcgagtcattcgtattttagtgtaccgtct-3’,如seqidno:9所示。
2、第二pcr扩增:以实验室保存的pet28a质粒为模板,f3/r3为引物进行载体pcr扩增,获得pet28a载体的线性片段,结果见图2中2泳道,引物设计如下:
f3:5’-tgactcgaggatccggctgct-3’,如seqidno:10所示;
r3:5’-ggtatatctccttcttaaagttaaacaaaa-3’,如seqidno:11所示。
3、将所述第一pcr扩增和所述第二pcr扩增获得的cl7-linker-his、3c-cvn和pet28a载体的三个线性片段用t5核酸外切酶在冰水浴中进行同源序列消化5min,产物进行常规转化大肠杆菌dh5a感受态细胞,挑取单菌落培养进行菌液pcr(图3)和质粒鉴定(图4),筛选阳性克隆,以得到pet28a-cl7-linker-his-3c-cvn的重组质粒,cl7-linker-his-3c-cvn的融合基因序列如seqidno:2所示,图3中3泳道为pcr鉴定的阳性克隆,图4中3泳道为质粒鉴定的pet28a-cl7-cvn的阳性重组质粒。同时构建了pet28a-cvn质粒作为对照。
上述引物中,f1(5’端的agaaggagatatacc)和r3(5’端的ggtatatctccttct)为同源序列,f2(5’端的agtcatcaccatcac)和r1(5’端的gtgatggtgatgact)为同源序列,f3(5’端的tgactcgaggatccg)和r2(5’端的cggatcctcgagtca)为同源序列,经过第一pcr扩增和第二pcr扩增得到的三个片段,通过t5核酸外切酶进行同源系列消化,在大肠杆菌dh5a感受态细胞内进行重组结合成环状,构建表达载体pet28a-cl7-cvn。
重组cl7-cvn蛋白的制备
1、将构建好的pet28a-cl7-cvn表达质粒转化至第一宿主细胞中,如大肠杆菌rosetta(de3)(merck公司);
2、进行诱导表达,iptg浓度0.5mmol/l,诱导温度为18℃,诱导表达时间16h;
3、离心收集诱导后的大肠杆菌,超声破碎,收集上清液,将收集的上清液于80℃水浴锅加热30min,离心收集获得上清;
4、将热处理后的上清液与ni-nta室温结合30-60min后,用0~30mm的咪唑洗涤镍珠,除去非特异性结合的杂蛋白;
5、用200-300mm的咪唑洗涤目的蛋白cl7-cvn;
6、将获得的目的蛋白用0.22um滤膜过滤除菌,分装低温保存;
7、一步纯化结果如图5,cl7-cvn纯度可达99%。
im7-beads的制备
im7蛋白的制备:将im7的表达载体pet-im7转化至第二宿主细胞中,如大肠杆菌bl21(de3)(merck公司)中,诱导表达im7蛋白;将诱导表达的细胞裂解上清于80℃水浴锅加热30min,离心收集上清,ni-nta一步亲和纯化,纯化结果见图6,纯度可达99%。
beads的制备:菊糖配制成溶液,加入六偏磷酸钠,充分搅拌溶解;再向体系中滴加碳酸钠溶液,调节ph为10-10.5,温度为40-50℃,搅拌反应3.5-4.0h,反应结束后调节ph至中性,用95%乙醇溶液沉淀多次,离心得到沉淀,沉淀用去离子水充分分散,得到beads分散液;其中,六偏磷酸钠占菊糖的投入质量百分比为8%-10%(记为shmp%)。
im7-beads的制备:取乳化剂,充分搅拌后,为油相液;将上述实施例纯化的im7蛋白、及beads分散液分别逐滴加入上述油相液中,乳化40-60min后;将pc溶液(花青素)逐滴加入该乳液中,37℃水浴,搅拌反应40-48h,反应结束后离心收集沉淀,用异丙醇洗涤三次,真空干燥,得到im7-beads微球。其中,im7、beads和pc的结合质量比(200-300):(100-150):(0.02-0.025),并且im7溶液、beads分散液和pc溶液的体积相等。上述乳化剂选自2%质量比的span-80溶液、或2%质量比的吐温-80溶液。
粒径跨度测定:以激光粒度分析仪测定微球的粒径,取干燥的微球样品悬浮于去离子水中,以超声分散。在样本接受管中,以2500rpm揽拌,并以50mv超声处理悬浮液30min,遮光率控制在5%-13%之间。
粒径跨度指数(微球粒径的范围):span=(d90-d50)/d10;
式中,d90、d50、d10和代表了微球粒径的第90、50和10百分位数,粒径的跨度指数代表了粒径的分布范围,每个样本重复三次测量。
取干燥的wd=0.1g微球悬浮于装有的5ml去离子水离心管中,在摇床上以中温和振动,然后每隔1h离心取样一次,用滤纸擦拭溶胀微球表面的水分,立即称重质量记为ws,溶胀率公式如下:esw(%)=[(ws-wd)/wd]*100%;
平衡水含量公式如下:ewc(%)=[(wc-wd)/wd]*100%;
wc为溶胀的微球在平衡水含量时的质量。
通过上述方式,本实施方式中共制备了10种微球,包括实施例1-6,和对比例1-4,其过程参数和微球的性能见表1。表1中列举了六偏磷酸钠占菊糖的投入质量百分比(shmp%),步骤s1的滴加的碳酸钠溶液用于调节ph至10-10.5,反应温度控制为40-50℃,反应时间为3.5-4.0h;im7、beads和pc的结合质量比,所述s2步骤中,乳化时间为40-60min后;将pc溶液逐滴后的反应时间40-48h;im7-beads粒径跨度(span),溶胀滤(esw,10h)和平衡水含量(ewc)。
其中,对比例4采用现有的琼脂糖微球和im7偶联,得到微球。具体方法为:将纯化的im7蛋白用缓冲液超滤换液,并浓缩至50mg/ml;用缓冲液洗涤琼脂糖微球,im7蛋白与微球室温混合结合15min,室温静置30min;50mm半胱氨酸避光封闭微球15min,静置30min后,用1mnacl洗涤微球3次,最后用nan3保存;
表1
由表1可以看出,实施例1-6相对于对比例1-4,其im7-beads平均粒径更小,其粒径跨度更小,其溶胀率更高,说明本发明实施例提供的im7-beads微球能够提供更优异的微观结构,为其富集和分离病毒提供更佳的效果。
elisas检测cl7-cvn、及cvn分别与prv病毒的结合能力
1、将感染prv病毒的pk15细胞,冻融3次;
2、将细胞裂解物,10000g离心10min,收集病毒上清;
3、elisas板用包被液包被病毒上清4℃,过夜;
4、用含有2%bsa的pbs在37℃封闭1h;
5、分别加入倍比稀释的cl7、cvn、cl7-cvn于37℃孵育1h;
6、用anti-his-taghrp的抗体检测结合的cvn、cl7-cvn;
7、加入100μl的1mhcl终止反应,酶标仪读取od450值。
结果见图7,随着稀释倍数的增加,od450值逐渐在减小,并且cl7-cvn与cvn实验组od450值高于cl7对照组,说明cl7-cvn与cvn都具有结合prv的活性,而cl7没有结合prv的活性。
im7-beads微球吸附病毒实施例的荧光强度观察
1、取等摩尔(0.4umol)的cl7,cvn,cl7-cvn分别与100μlprv(gfp标记,106viruses/μl)病毒液进行混合孵育30min;再分别加入50μl的im7-beads微球,室温混合孵育30min;其中im7-beads微球采用上述实施例5制备的微球;
2、离心分离上清和微球,微球用couplingbuffer(0.1mtris-hcl,ph8.0,0.5mnacl)/1mnacl洗涤3-5次;
3、吸附后的结果用蓝光仪观察荧光强度。
结果如图8,ep管4,6,8中,洗涤后的微球没有检测到绿色荧光信号,说明分别加入pbs,cl7,cvn不能使im7-beads结合吸附prv;而ep管9中,通过加入im7-beads、及重组cl7-cvn蛋白,在ep管10中发现洗涤后的微球出现绿色荧光,由于prv病毒在蓝光照射下能够产生荧光,说明im7-beads能通过重组cl7-cvn蛋白与囊膜病毒的识别结合,实现囊膜病毒的分离富集。im7-beads微球、重组cl7-cvn蛋白和病毒的吸附原理如图1所示。
rt-pcr初步检测prv病毒的分离富集
1、取等摩尔(0.4μmol)的cl7,cvn,cl7-cvn分别与100μl的prv病毒液(gfp标记,106viruses/μl)混合室温孵育30min;再分别加入50μl的im7-beads,室温混合孵育30min;其中im7-beads微球采用上述实施例5-6,及对比例1-4制备的微球;
2、离心分离上清和微球,微球用couplingbuffer(0.1mtris-hcl,ph8.0,0.5mnacl)/1mnacl洗涤3-5次;
3、分别提取上清和微球的病毒基因组;荧光定量pcr法(rt-pcr)检测富集效率。
4、富集效率用下式表示:
结果如图9,pbs、cl7和cvn均不能使im7-beads有效实现病毒的分离和富集;而cl7-cvn能使im7-beads实现prv病毒的分离和富集,富集效率高达90.78%;并且实施例5-6制备的im7-beads微球用来吸附病毒,其富集效率均显著高于对比例1-4,如图10所示。
pcr法验证prv病毒的分离富集检测
1、分别将pbs溶液、cl7蛋白溶液、cvn蛋白溶液和cl7-cvn蛋白溶液与100μlprv病毒液(gfp标记,106viruses/μl)混合室温孵育30min;
2、再分别加入im7-beads微球,室温混合孵育30min;其中im7-beads微球采用上述实施例5制备的微球;
3、静置或简单离心让微球沉降分离;微球用couplingbuffer(0.1mtris-hcl,ph8.0,0.5mnacl)/1mnacl洗涤3-5次;
4、洗涤微球后,分别提取微球的prv基因组,pcr分别检测gd基因的片段(226bp),gb基因片段(331bp);计算富集效率。
结果如图11,cl7-cvn的处理组都能检测到gd基因的片(226bp),gb基因片段(331bp),而pbs、cl7和cvn的处理组都没有检测到;再次印证仅cl7-cvn能使im7-beads实现prv病毒的分离和富集。
不同浓度的cl7-cvn对prv病毒富集效率的影响
1、分别将不同摩尔量的cl7-cvn(0,0.125,0.25,0.5,1μmol)与100μl的prv病毒液(gfp标记,106viruses/μl)混合室温孵育30min;
2、再分别加入50μl的im7-beads,室温混合孵育30min;其中im7-beads微球采用上述实施例5制备的微球;
3、静置或离心让微球沉降分离;
4、分别提取上清和微球的病毒基因组;rt-pcr检测富集效率;
结果如图12,cl7-cvn的处理浓度为0.125-1μmol之间,对应的50μl的im7-beads,即为,cl7-cvn结合im7-beads的处理浓度为2.5-20μmol/mlim7-beads微球,对应的吸附100μl的病毒液浓度为106viruses/μl;当cl7-cvn处理浓度为0.5μmol时,即cl7-cvn结合im7-beads的处理浓度为10μmol/mlim7-beads微球时,此时富集效率可达97.32%,即为最佳处理浓度。
将prv(gfp标记)病毒样本分离富集后的上清去感染细胞分析
1、取等量的病毒样本与结合cl7-cvn的im7-beads混合孵育;
2、同时设置对照,正常细胞,没有富集分离的病毒感染细胞;
3、离心收集上清病毒液,过滤除菌;
4、将除菌后的病毒液分别感染细胞;
5、培养16h荧光显微镜观察病毒感染情况;
结果如图13,通过细胞感染的情况,表明病毒样本经过im7-beads与cl7-cvn处理吸附后,病毒样本上清中病毒粒子明显减少,表明采用im7-beads和重组cl7-cvn能实现对prv病毒的分离和富集,为实际对生物体病毒感染的检测提供依据。
富集prv病毒的敏感性
1、分别将100μlprv病毒(106viruses/μl)稀释到终体积200、400、800、1000μl;
2、将0.5μmol的cl7-cvn蛋白与不同体积的prv病毒液分别室温孵育30min;
3、再分别加入50μl的im7-beads,室温混合孵育30min;
4、静置或简单离心让微球沉降分离;
5、分别提取上清和微球的病毒基因组,采用rt-pcr检测富集效率;
6、pcr检测微球富集的prvgk基因片段;
rt-pcr检测富集效率结果如图14a,当100μlprv病毒液(106virus/μl)稀释到1ml时(浓度为105viruses/μl),采用0.5μmol的重组cl7-cvn蛋白和50μl的im7-beads,其富集效率依然能够达到88.72%,说明本发明提供的分离富集prv方法的敏感度高,可为后面的基因检测铺垫;pcr检测gk基因片段结果如图14b,表明当样本稀释10倍,依旧能检测到prv的基因,进一步证实该富集方法的灵敏性较好,可用于大体积样本的富集检测。
富集prv病毒的容量
1、分别取106viruses/μl的prv病毒100、200、300、400、500μl;
2、将50μl结合1μmolcl7-cvn的im7-beads与不同体积的病毒液分别室温孵育30min;im7-beads微球采用上述实施例5制备的微球;cl7-cvn与im7-beads的结合比见表2;
3、静置或简单离心让微球沉降分离;
4、分别提取上清和微球的病毒基因组,采用rt-pcr检测富集效率,并计算富集容量,富集容量为每毫升im7-beads所能富集的病毒量。
图15,当106viruses/μl的prv病毒样本从100μl增加到300μl时,使用50μl结合1μmolcl7-cvn的im7-beads,病毒的富集量增加了3倍,im7-beads富集病毒的容量约为5.41×109viruses/ml,并且此时的病毒的富集效率达到了89.64%;病毒样本从300μl-500μl阶段,其富集效率和富集容量增加不大,说明其结合病毒接近于饱和。
表2中,采用106viruses/μl的prv病毒样本500μl时,以使得微球饱和吸附病毒,并考察了不同的cl7-cvn与im7-beads结合质量体积比(μmol/ml)、及孵育时间对富集效率和富集容量的影响。结果如表2所示,cl7-cvn与im7-beads结合的质量体积比为2.5-20μmol/ml,cl7-cvn|、im7-beads和病毒的总孵育时间为40-100min。当cl7-cvn和im7-beads质量比为5-15μmol/ml,总孵育时间为60-100min时,富集效率86.36%-93.16%,富集容量5.41×109-1.06×1010viruses/ml。且当cl7-cvn与im7-beads结合的质量体积比为10μmol/ml,孵育时间为30min时,富集效率和富集容量达到最大。
表2
综上所述:
1、本发明通过基因工程的方法制备了im7蛋白,并以此制备了im7-beads微球,im7-beads微球平均粒径为82.35±0.35μm至151.27±0.48μm之间,能够提供更优异的微观结构和溶胀性能,为其富集和分离病毒提供更佳的效果。
2、本发明通过基因工程的方法制备了重组cl7-cvn蛋白,并利用cl7与im7的特异性结合、及cvn结合囊膜病毒的性能,能够将prv病毒颗粒富集至im7-beads表面,为病毒分离富集和其检测提供一种全新的技术手段;并且im7-beads微球富集病毒的容量达到1.06×1010viruses/ml。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
序列表
<110>湖北大学
<120>基于cl7-cvn和im7系统分离、富集和检测囊膜病毒的方法
<160>9
<170>siposequencelisting1.0
<210>2
<211>771
<212>dna
<213>人工序列(artificialsequence)
<400>2
atgggcagcaaaagcaatgaaccgggtaaagcaaccggtgaaggtaaaccggttaataac60
aaatggctgaacaatgccggtaaagatctgggtagtccggttccggatcgtattgcaaat120
aaactgcgtgataaagaattcgagagcttcgatgattttcgtgaaaccttttgggaagaa180
gttagcaaagatcctgaactgagcaaacagtttagccgcaataacaatgatcgtatgaaa240
gttggtaaagcaccgaaaacacgtacccaggatgttagcggtaaacgtacctcatttgaa300
ctgaatcatcagaaaccgattgaacagaatggtggcgtttatgatatggataacattagc360
gttgttaccccgaaacgcaacattgatattgaaggcggtggcggagggagcggtggaggg420
ggcagtcatcaccatcaccatcacttggaggttttgttccagggtccacttggtaaattc480
tcccagacctgctacaactccgctatccagggttctgttctgacctctacctgcgaacgt540
accaacggtggttataacacctcctctatcgacctgaactccgttattgaaaacgttgac600
ggttctctgaaatggcagccatctaacttcatcgaaacctgccgtaacacccagctggct660
ggttcctctgaactggctgctgaatgcaaaacccgtgctcagcagttcgtttctaccaaa720
atcaacctagacgaccatatagctaacatagacggtacactaaaatacgaa771
<210>3
<211>396
<212>dna
<213>人工序列(artificialsequence)
<400>3
atgggcagcaaaagcaatgaaccgggtaaagcaaccggtgaaggtaaaccggttaataac60
aaatggctgaacaatgccggtaaagatctgggtagtccggttccggatcgtattgcaaat120
aaactgcgtgataaagaattcgagagcttcgatgattttcgtgaaaccttttgggaagaa180
gttagcaaagatcctgaactgagcaaacagtttagccgcaataacaatgatcgtatgaaa240
gttggtaaagcaccgaaaacacgtacccaggatgttagcggtaaacgtacctcatttgaa300
ctgaatcatcagaaaccgattgaacagaatggtggcgtttatgatatggataacattagc360
gttgttaccccgaaacgcaacattgatattgaaggc396
<210>4
<211>132
<212>prt
<213>人工序列(artificialsequence)
<400>4
metglyserlysserasngluproglylysalathrglygluglylys
151015
provalasnasnlystrpleuasnasnalaglylysaspleuglyser
202530
provalproaspargilealaasnlysleuargasplysglupheglu
354045
serpheaspaspphearggluthrphetrpglugluvalserlysasp
505560
progluleuserlysglnpheserargasnasnasnaspargmetlys
65707580
valglylysalaprolysthrargthrglnaspvalserglylysarg
859095
thrserphegluleuasnhisglnlysproilegluglnasnglygly
100105110
valtyraspmetaspasnileservalvalthrprolysargasnile
115120125
aspileglugly
130
<210>6
<211>31
<212>dna
<213>人工序列(artificialsequence)
<400>6
agaaggagatataccatgggcagcaaaagca31
<210>7
<211>60
<212>dna
<213>人工序列(artificialsequence)
<400>7
gtgatggtgatgactgccccctccaccgctccctccgccaccgccttcaatatcaatgtt60
<210>8
<211>61
<212>dna
<213>人工序列(artificialsequence)
<400>8
agtcatcaccatcaccatcacttggaggttttgttccagggtccacttggtaaattctcc60
c61
<210>9
<211>37
<212>dna
<213>人工序列(artificialsequence)
<400>9
cggatcctcgagtcattcgtattttagtgtaccgtct37
<210>10
<211>21
<212>dna
<213>人工序列(artificialsequence)
<400>10
tgactcgaggatccggctgct21
<210>11
<211>30
<212>dna
<213>人工序列(artificialsequence)
<400>11
ggtatatctccttcttaaagttaaacaaaa30
- 还没有人留言评论。精彩留言会获得点赞!