一种过氧麦角甾醇的制备与检识方法与流程
本发明属于医药技术领域。更具体地,涉及一种过氧麦角甾醇的制备与检识方法。
背景技术:
过氧麦角甾醇全称为3β-羟基-5α,8α-过氧化麦角甾-6,22-二烯,结构式如式(ⅰ)所示:
现有技术中制备过氧麦角甾醇的方法主要为先分离未知单体后鉴别过氧麦角甾醇,如专利201210233192.1利用酶法破壁孢子粉经超临界二氧化碳萃取,先后以氯仿、乙酸乙酯、石油醚-乙酸乙酯混合溶剂、石油醚-丙酮混合溶剂等为洗脱剂,经3次正向硅胶柱分离,最终再采用制备色谱精制获得活性最高的单体成分,鉴别为过氧麦角甾醇。专利200610126091.9以簇生沿丝伞子实体或菌丝体为原料,通过甲醇提取,石油醚萃取,上正向硅胶柱,环己烷-乙酸乙酯混合溶剂洗脱,合并相同流分,经多次重结晶获得两个单体,其中一个鉴别为过氧麦角甾醇。
然而,这种从未知出发先获得单体,再鉴定出是过氧麦角甾醇容易出现误判,如章能胜等(玫烟色拟青霉中过氧麦角甾醇的高速逆流色谱法分离纯化及质谱分析,食品与发酵工业,2009,35(6):14-16.)采用较为先进的高速逆流色谱法对玫烟色拟青霉中过氧麦角甾醇进行分离纯化,但其获得的目标产物并非是真正的过氧麦角甾醇,因为过氧麦角甾醇在240-400nm无紫外吸收,而文中分离过程的监视和目标物的纯度检测均采用280nm紫外吸收波长,且在色谱图中均显示出较大的吸光度,可能是作者误判。
上述摸索式分离的制备方法不仅容易出现误判,造成人力与物料的浪费,并且制备工艺复杂,不适合工业化生产。
技术实现要素:
针对现有技术存在的技术缺陷,本发明的目的是提供一种新的过氧麦角甾醇制备方法,通过先对流份中过氧麦角甾醇精准检识,再根据检识结果提取过氧麦角甾醇,该种对过氧麦角甾醇的针对性提取方法能大大降低错误率,避免盲目性,并且整个过程简便易行,适合工业化生产。
本发明的目的是提供一种过氧麦角甾醇的制备方法。
本发明另一目的是提供一种过氧麦角甾醇的检识方法。
本发明另一目的是提供过氧麦角甾醇在制备抗肿瘤药物及降血糖药物中的应用。
本发明上述目的通过以下技术方案实现:
一种过氧麦角甾醇的制备方法,包括以下步骤:
(1)流份制备:将兰科开唇兰属植物的干药材粉碎,乙醇提取浓缩、有机溶剂萃取得浸膏,通过层析柱洗脱得流份;
(2)过氧麦角甾醇的检识:对所述流份进行hptlc检识和/或uhplc-esi-ms检识;
(3)过氧麦角甾醇的精制:根据步骤(2)的检识结果,选择含有过氧麦角甾醇的流份合并,经过柱层析后重结晶得到过氧麦角甾醇成品。
优选地,所述兰科开唇兰属植物包括小片齿唇兰、短柱齿唇兰、滇南开唇兰、白齿唇兰、滇越金线兰、红萼齿唇兰、小齿唇兰、西南齿唇兰、峨眉金线兰、台湾银线兰、耿马齿唇兰、台湾齿唇兰、恒春银线兰、齿唇兰、艳丽齿唇兰、屏边金线兰、金线兰、保亭金线兰(变种)、一柱齿唇兰、香港金线兰、浙江金线兰。
本发明利用hptlc检识及uhplc-esi-ms检识技术建立系统化的快速检识过氧麦角甾醇的方法,首先对流份中过氧麦角甾醇进行精确识别,然后选择含有过氧麦角甾醇的流份合并提取,大大提高制备效率,避免制备过程中的盲目性,从而避免人力与物料的浪费。
优选地,步骤(2)中hptlc检识的显色剂为10%硫酸乙醇显色剂和/或1,1-二苯基-2-三硝基苯肼显色剂。
优选地,1,1-二苯基-2-三硝基苯肼(dpph)显色剂的浓度为15mg/100ml,以甲醇为溶剂。
本发明通过对hptlc显色剂的选择,使得hptlc检测方法呈现出过氧麦角甾醇的专属性以及稳定性,使得检测结果更加准确。
优选地,步骤(2)中hptlc检识的展开剂为石油醚/乙酸乙酯、氯仿/丙酮、氯仿/甲醇中的一种或多种。
优选的,石油醚∶乙酸乙酯为2∶1、氯仿∶丙酮为15∶1以及氯仿∶甲醇为20∶1作为展开剂。
优选地,步骤(2)中uhplc-esi-ms检识的色谱条件为色谱柱为shim-packxr-odsⅱcolumn(100mm×2.0mm,2.2μm),流速为0.3-1ml/min,流动a相为甲醇,流动b相为蒸馏水,用梯度洗脱模式,0-20min流动a相从10%梯度洗脱至100%流动a溶液,进样量为10-20μl。
更优选地,流速为0.3ml/min,梯度洗脱模式为:0-20min流动a相从10%梯度洗脱至100%流动a溶液,进样量为10μl。
优选地,步骤(2)中uhplc-esi-ms检识的质谱条件为离子源选择电喷雾离子源,在正离子模式或负离子模式测定所述流份。
优选地,步骤(2)中uhplc-esi-ms检识还包括质谱数据处理的过程,具体为:输入过氧麦角甾醇分子式后,查找存在相同分子量碎片的液相峰,分别解析各个液相峰所对应的质谱图,最终确定目标化合物所对应的液相峰。
通过筛选uhplc-esi-tofms检识条件可有效地从复杂成分中检识出过氧麦角甾醇且无需标准品。
优选地,步骤(3)中柱层析中层析柱为sephadexlh-20,洗脱剂为正己烷/氯仿/甲醇、氯仿/甲醇、纯甲醇中的一种或多种。
更优选地正己烷:氯仿∶甲醇为2∶1∶1、氯仿∶甲醇为1∶1。
一种过氧麦角甾醇的检识方法,所述方法为对流份进行hptlc检识和/或uhplc-esi-ms检识;其中,hptlc检识中显色剂为10%硫酸乙醇显色剂和/或1,1-二苯基-2-三硝基苯肼显色剂;其中,uhplc-esi-ms检识中色谱条件为色谱柱为shim-packxr-odsⅱcolumn(100mm×2.0mm,2.2μm),uhplc-esi-ms检识的色谱条件为色谱柱为shim-packxr-odsⅱcolumn(100mm×2.0mm,2.2μm),流速为0.3-1ml/min,流动a相为甲醇,流动b相为蒸馏水,用梯度洗脱模式,0-20min流动a相从10%梯度洗脱至100%流动a溶液,进样量为10-20μl;质谱条件为离子源选择电喷雾离子源,在正离子模式或负离子模式测定所述流份。
更优选地,流速为0.3ml/min,进样量为10μl。
本发明通过大量实验筛选hptlc显色剂发现,10%硫酸乙醇显色剂与1,1-二苯基-2-三硝基苯肼显色剂均能检识过氧麦角甾醇,并且1,1-二苯基-2-三硝基苯肼显色剂效果优于10%硫酸乙醇显色剂,且二者联用显示效果及持久性优于单独使用。通过筛选uhplc-esi-ms检识条件后能有效地从复杂成分中检识出过氧麦角甾醇,并且将hptlc检识结合uhplc-esi-ms检识能提高过氧麦角甾醇的检识精确度。
过氧麦角甾醇在制备用于抗肿瘤药物中的应用,其中,所述抗肿瘤药物为治疗肺癌、食道癌或胃癌药物中的一种或多种。
过氧麦角甾醇在制备用于降血糖药物中的应用。
本发明具有以下有益效果:
本发明利用hptlc与uhplc-esi-ms技术对粗品流份进行过氧麦角甾醇检识,并筛选hptlc显色剂与uhplc-esi-ms检识条件,提高了过氧麦角甾醇检识的精准度,整个检识过程速度快、稳定性高。根据准确的检识结果再针对性提取过氧麦角甾醇,能有效避免提取失误情况,提高制备效率,避免物料及人力的浪费。
本发明首次将过氧麦角甾醇应用于治疗肺癌、食道癌或胃癌,结果表明,过氧麦角甾醇能抑制肺癌、食道癌、胃癌细胞生长,其中过氧麦角甾醇浓度为40-80μmol/l对食道癌、胃癌细胞生长效果明显,60-80μmol/l对肺癌细胞生长效果明显,表明过氧麦角甾醇可以作为在制备治疗肺癌、食道癌、胃癌药物中的应用。另外,本发明首次将过氧麦角甾醇应用于降血糖治疗,实验结果表明高剂量过氧麦角甾醇给药组葡萄糖摄取量是模型组的1.20倍,过氧麦角甾醇可较好地改善胰岛素抵抗。因此过氧麦角甾醇可作为在制备降血糖药物中的应用。
附图说明
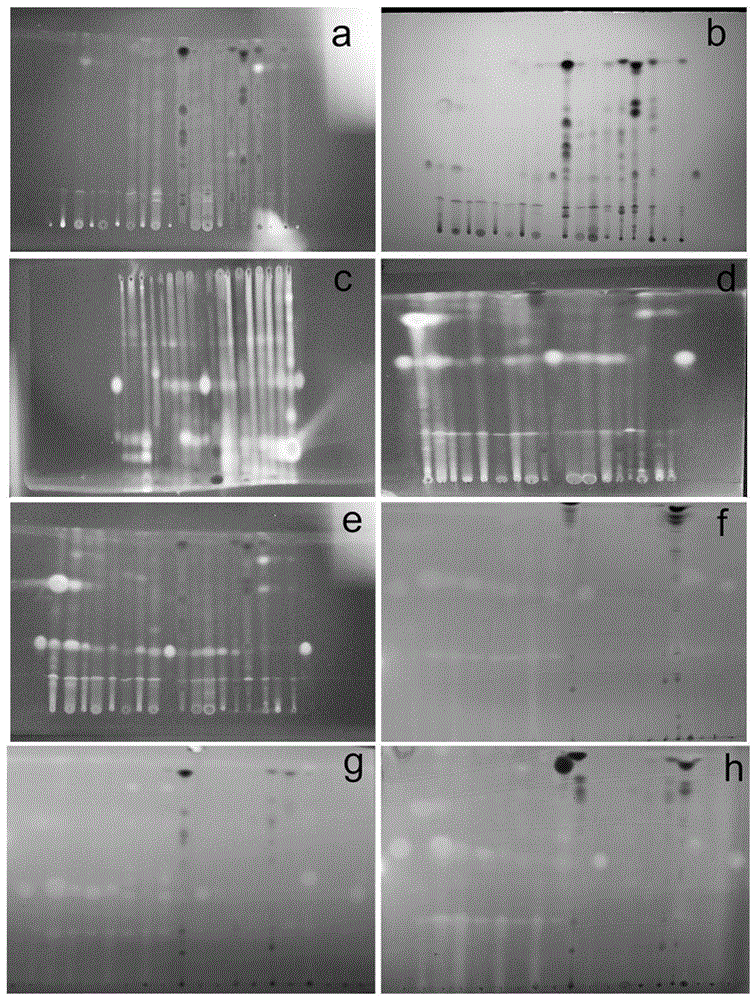
图1为hptlc检识中hptlc色谱,图(a)甲醇∶氯仿1∶20在365nm紫外光下;(b)甲醇∶氯仿1∶20用10%硫酸乙醇染色后在可见光下;(c)石油醚∶乙酸乙酯2∶1用10%硫酸乙醇染色后在365nm紫外光下;(d)丙酮∶氯仿1∶15用10%硫酸乙醇染色后在365nm处紫外光下;(e)甲醇∶氯仿1∶20用10%硫酸乙醇染色后在365nm紫外光下;(f)丙酮∶氯仿1∶15用dpph染色后在可见光下;(g)甲醇∶氯仿1∶20用dpph染色后在可见光下;(h)丙酮∶氯仿1∶15用10%硫酸醇-dpph染色后在可见光下;
图2为质谱总离子流图,(a)正离子模式;(b)负离子模式;
图3为质谱总离子流图中碎片离子筛选图,(a)正离子模式下的筛选条件是[c28h44o3+h]+;(b)正离子模式下的筛选条件是[c28h44o3+na]+;(c)负离子模式下的筛选条件为[c28h44o3-h]-;
图4为在正离子模式下,筛选条件是[c28h44o3+na]+的质谱总离子流图,(a)88-92流份;(b)124-133流份;(c)石油醚部位;(d)氯仿部位;
图5为质谱图,(a)80-84在保留时间12.704min所相对应液相峰的质谱图;(b)80-84在保留时间11.802min所相对应液相峰的质谱图;
图6为三个层析体系共薄层图,(a)石油醚∶乙酸乙酯2∶1;(b)甲醇∶氯仿20∶1;(c)丙酮∶氯仿15∶1;
图7为sgc-7901经ao/eb染色后荧光显微镜图,(a)空白组;(b)20μmol/l剂量组;(c)40μmol/l剂量组;
图8为sgc-7901细胞凋亡率图,(a)空白组;(b)20μmol/l剂量组;(c)40μmol/l剂量组;(d)60μmol/l剂量组;
图9为过氧麦角甾醇对sgc-7901细胞周期的影响,(a)空白组;(b)20μmol/l剂量组;(c)40μmol/l剂量组;(d)60μmol/l剂量组;
图10为荧光显微镜下染色后的sgc-7901癌细胞活性氧情况,(a)空白组;(b)rosup阳性组;(c)20μmol/l剂量组;(d)40μmol/l剂量组;(e)60μmol/l剂量组;
图11为sgc-7901癌细胞活性氧定量;
图12为荧光显微镜下观察sgc-7901癌细胞经jc-1染色后的情况;
图13为各组sgc-7901癌细胞线粒体膜电位定量。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
实施例1过氧麦角甾醇的检识
1、流份制备:将小片齿唇兰的干药材粉碎,将其置于提取罐中,使用10倍量的95%乙醇在50℃下提取三次,再将其在70℃下提取三次,每次提取3h,将依次过滤得到的提取液在45℃浓缩罐中浓缩得到总浸膏。将浸膏混悬于适量的蒸馏水中,分别用石油醚、氯仿、乙酸乙酯以及正丁醇多次萃取浓缩得石油醚部位浸膏、氯仿部位浸膏、乙酸乙酯部位浸膏、正丁醇部位浸膏以及水部位浸膏。将石油醚部位浸膏以样品量∶硅胶量1∶1.5比例拌样制成硅胶干粉,以样品量与硅胶量1∶30装填硅胶层析柱,每1500ml作为一个流份,分别通过纯石油醚、石油醚∶乙酸乙酯50∶1-1∶10以及纯乙酸乙酯作为洗脱液梯度洗脱,共得到491个流份,点板将相似流份合并。
2、过氧麦角甾醇检识过程:
(1)hptlc检识过程:
显色剂配制:硫酸乙醇显色剂:用量筒量取10ml浓硫酸加于100ml无水乙醇中充分搅拌均匀后,室温下放置备用;dpph显色剂:用分析天平称量1,1-二苯基-2-三硝基苯肼(dpph)15mg溶于100ml甲醇中,充分搅拌溶解均匀后放置,dpph溶液不稳定,需要现配现用,存放于阴凉处。
点样跑板:分别从各萃取部位以及硅胶层析的流份中取出适量样品置于10ml西宁瓶中加适量的溶剂进行溶解,用毛细管吸取依次将样品点于高效薄层板中,分别以石油醚∶乙酸乙酯为2∶1、氯仿∶丙酮为15∶1以及氯仿∶甲醇为20∶1作为展开剂层析,放置晾干。
显色:将晾干的高效薄层板分别用10%硫酸乙醇、dpph溶液及其10%硫酸乙醇与dpph溶液联用进行显色,将10%硫酸乙醇显色的高效薄硅胶层板置于95℃烘箱中15min后取出观察,将dpph溶液和两者显色剂联用显色的高效薄层硅胶板放置室温光照条件下10min后取出观察。
(2)uhplc-esi-ms检识过程:
样品处理:根据hptlc检识结果,将具有相同斑点的流份合并后,选择含有过氧麦角甾醇的流份80-84、88-92、124-133、石油醚部位浸膏、氯仿部位浸膏作为样品。使用分析天平分别称定80-84、88-92、124-133、石油醚部位浸膏以及氯仿部位30mg于西宁瓶中,溶于4ml色谱甲醇后超声后,分别吸取西宁瓶样品上清液1ml,经0.22μm微孔滤膜过滤后置于进样瓶备用。
测定条件:色谱条件:色谱柱为shim-packxr-odsⅱcolumn(100mm×2.0mm,2.2μm),流速为0.3ml/min,流动a相为色谱甲醇,流动b相为蒸馏水,用梯度洗脱模式,0-20min流动a相从10%梯度洗脱至100%流动a溶液,进样量为10μl;质谱条件:离子源选择电喷雾离子源(esi),分别在正、负离子模式测定样品。
数据处理:使用peakview软件(absciex)进行质谱数据处理,输入已知化合物的分子式后,通过formulafinder的功能精确查找存在相同分子量碎片的液相峰,从而除去杂质干扰峰确定目标化合物所可能对应的液相峰,分别解析各个液相峰所对应的质谱图,最终确定目标化合物所对应的液相峰。
3、过氧麦角甾醇检识结果与分析:
(1)hptlc检识结果与分析:
在图1中,从左到右的点如下:过氧麦角甾醇标准品、74-79、80-84、85-87、88-92、93-106、107-121、122-123、124-133、134-151、过氧麦角甾醇标准品、152-155、156-170、171-207、208-222、223-236、石油醚部位、乙酸乙酯部位、氯仿部位和过氧麦角甾醇标准品。图1a中可发现未经显色的高效薄层板在紫外下无明显的目标斑点,这验证了过氧麦角甾醇不具有长共轭结构,存在紫外弱吸收的特点。从图1b、1c、1d、1e可以看出,将高效薄层板通过10%硫酸乙醇显色后在可见光下虽然可以看到斑点但相对较弱。在紫外灯波长365nm下,可以清晰地观察到目标斑点。在高效薄层板中计算得出在三种层析体系乙酸乙酯∶石油醚1∶2、甲醇∶氯仿1∶20以及丙酮∶氯仿1∶15中目标化合物的rf值分别为0.51、0.47、0.64,结果表明流份74-222均存在过氧麦角甾醇,但由于hptlc检识灵敏性不足无法准确检识出石油醚层浸膏、氯仿层浸膏、乙酸乙酯层浸膏是否含有过氧麦角甾醇。从图1f、1g研究发现,使用dpph作为显色剂,可以有效地避免杂质点的干扰,有利于目标化合物的检识。
10%硫酸乙醇-dpph显色剂联用(图1h)增强了检识实验的稳定性,通过实验证明了10%硫酸乙醇-dpph显色剂联用后显色斑点持续时间比单独使用dpph显色明显延长。将流份80-84、85-87、88-92、93-106与对照品点样跑板后分别使用dpph和10%硫酸乙醇-dpph显色,进行三次平行实验。实验中用dpph显色的高效薄层板斑点在3h后出现褪色现象,4h斑点完全消失,如光照较弱时其褪色速度更为快速,而10%硫酸乙醇-dpph显色的薄层板在5d后仍然存在明显的斑点。
(2)uhplc-esi-ms检识结果与分析:
将80-84样品自动进样,通过液相色谱柱梯度洗脱进行成分分离,经过质谱检测器,在正离子与负离子模式下得到80-84的总离子质谱图分别为图2a、2b。在图2中可知流份80-84含有较多的化合物,需要通过分析软件筛选出目标化合物液相峰。通过peakview中formulafinder功能搜寻,使图2a中只显示含有与c28h44o3相同相对分子量离子的液相峰,分别以[c28h44o3+h]+分子量(429.6470)和[c28h44o3+na]+分子量(451.6470)为搜寻条件得到图3a、3b。从图3a中可以看出具有相对分子量为429.6470碎片离子的液相峰存在一个明显的大峰与些许小峰,经过对各个峰所对应的质谱进行解析都无法从中找出过氧麦角甾醇所对应的峰,从中可以得出过氧麦角甾醇[c28h44o3+h]+的质谱响应值较小,无法寻找出目标峰。从图3b可以发现具有相对分子量为451.6470碎片离子的液相峰存在两个大峰与些许的小峰,通过对各个峰所对应质谱的解析发现,在其12.704min所对应的液相峰便是目标液相峰,从中可以得出[c28h44o3+na]+准分子离子中esi源中响应值较大,便于对过氧麦角甾醇的检出。在负离子模式下80-84总离子质谱图搜寻含有[c28h44o3-h]-相同相对分子量碎片离子的液相峰得到图3c,图中含有与检索目标相对分子量相同的液相峰较多,不利于检识。综上所得,uhplc-esi-tofms检识过氧麦角甾醇的检识条件为在正离子模式下,以[c28h44o3+na]+准分子离子的相对分子量为检索条件。
通过各图谱的对比决定选用各正离子模式总离子流色谱图,且选用[c28h44o3+na]+作为筛选条件,对各个样品总离子流质谱图进行处理后得到图4a、4b、4c、4d。分别对图3b的2个大峰进行解析,对保留时间12.704min所对应峰的质谱图进行数据处理评估得到表1,实际相对分子量与测量相对分子量比值所得出的相对偏差值为-3.7ppm位于允许误差±5.0ppm之内。保留时间12.704min所对应峰的质谱图5a中存在一个相对分子量为429.3345的质谱峰,可归属为[m+h]+准分子离子峰。相对分子量为451.3161的质谱峰与[m+na]+准分子离子峰相一致,相对分子量为879.639的质谱峰与[2m+na]+准分子离子峰相一致,两个峰均具备响应值较大的特点,这与过氧麦角甾醇的质谱表现相一致。而相对分子量为396.3340的质谱峰归属为[m-o2]+离子碎片峰,这是过氧键(-o-o-)断裂后产生的碎片离子,[m-o2]+为过氧麦角甾醇特征性碎片离子。虽然保留时间为11.802min处峰所对应的质谱图(图5b)具有[m+na]+准分子离子的特征峰,但在质谱图中并没有找到相对应的[m+h]+、[2m+na]+、[m-o2]+质谱峰,并且图中存在高响应值的相对分子量为611.4227的质谱峰无法被归属,综上所述保留时间为12.704min所对应的液相峰可以鉴定为化合物过氧麦角甾醇液相色谱峰。
按照上述的方法分别对于样品80-84、88-92、124-133、石油醚部位及其氯仿部位所对应的液相图以及质谱图进行分析,实验得出80-84、88-92、124-133及其石油醚部位均含有过氧麦角甾醇,而氯仿部位含量较少。
表1质谱数据分析
(3)化合物结构解析及鉴定:
esi-msm/z:429.33295[m+h]+,879.64047[2m+na]+;mp.181-183℃;1h-nmr(500mhz,cdcl3)δ:0.81(3h,d,j=2.3hz),0.83(3h,s),0.84(3h,d,j=2.1hz),0.88(3h,s),0.91(3h,dd,j=6.8,3.5hz),1.0(3h,d,j=6.6hz)可以得出该化合物具有6个甲基,3.98(1h,m)是羟基所连碳上氢质子的化学位移,5.14(1h,dd,j=15.3,8.3hz),5.22(1h,dd,j=15.3,7.5hz)为双键两侧碳上的2个氢质子化学位移,6.24(1h,d,j=8.5hz),6.50(1h,d,j=8.5hz)为双键两侧碳上的2个氢质子化学位移;在13c-nmr(125mhz,cdcl3)中显示该化合物具有28个碳原子,化学位移值δ135.41,130.75为双键上两个碳原子的化学位移,δ135.20,132.31为化合物另一个双键结构上两个碳原子的化学位移,δ82.17,79.44为氧原子所连季碳上的化学位移值,此为过氧键的信号峰,δ66.48显示该化合物具有羟基,13c-nmr还显示出该化合物具有多个甲基、亚甲基以及次甲基,δ56.19(c-17),51.68(c-14),51.08(c-9),44.56(c-13),42.77(c-24),39.74(c-20),39.34(c-12),36.96(c-10),36.91(c-4),34.69(c-1),33.07(c-25),30.10(c-2),28.65(c-16),23.40(c-11),20.88(c-21),20.63(c-15),19.95(c-27),19.64(c-26),18.18(c-19),17.56(c-28),12.87(c-18)。1h-nmr以及13c-nmr数据与文献报道基本一致。将样品与过氧麦角甾醇对照品进行共薄层分析,将在三个不同体系层析后的hptlc通过10%硫酸乙醇显色后得到图6,薄层板上的三个点依次为样品斑点、二者叠加的斑点、对照品斑点。从图6a、6b、6c中可以看出样品与过氧麦角甾醇对照品rf一致,故确定该化合物为3β-羟基-5α,8α-过氧化麦角甾-6,22-二烯。
实施例2过氧麦角甾醇的检识
1、流份制备:同实施例1。
2、过氧麦角甾醇检识:采用hptlc检识各流份,检识方法同实施例1中hptlc检识过程。
实施例3过氧麦角甾醇的检识
1、流份制备:同实施例1。
2、过氧麦角甾醇检识:采用uhplc-esi-ms检识各流份。
样品处理:同实施例1。
测定条件:色谱条件:色谱柱为shim-packxr-odsⅱcolumn(100mm×2.0mm,2.2μm),流速为1ml/min,流动a相为色谱甲醇,流动b相为蒸馏水,用梯度洗脱模式,0-20min流动a相从10%梯度洗脱至100%流动a溶液,进样量为20μl;质谱条件:离子源选择电喷雾离子源(esi),分别在正、负离子模式测定样品。
数据处理:使用peakview软件(absciex)进行质谱数据处理,输入已知化合物的分子式后,通过formulafinder的功能精确查找存在相同分子量碎片的液相峰,从而除去杂质干扰峰确定目标化合物所可能对应的液相峰,分别解析各个液相峰所对应的质谱图,最终确定目标化合物所对应的液相峰。
实施例4过氧麦角甾醇的制备
1、流份制备:同实施例1。
2、过氧麦角甾醇检识:同实施例1。
3、过氧麦角甾醇提取:
在步骤2检识结果的基础上,选择含有过氧麦角甾醇的流份以过氧麦角甾醇斑点为主要观察对象通过hptlc-dpph的方法将含有相同斑点的流份合并。将含有过氧麦角甾醇的流份107-121、122-123、124-133、134-151、152-155、156-170、171-207以及208-222合并富集得到浸膏41.15g。将样品∶硅胶为1∶1.5的比例制成硅胶干粉,以样品∶硅胶为1∶50的比例进行硅胶柱层析,洗脱液为石油醚∶乙酸乙酯=15∶1-1∶10,将得到的流份通过hptlc-dpph的方法进行目标成分跟踪,合并后得到组分1。将流份74-79、80-84、85-87、88-92、93-106以及组分1分别通过sephadexlh-20柱层析,其洗脱液体系为正己烷∶氯仿∶甲醇=2∶1∶1,将得到的流份进行目标成分跟踪合并得到组分2。组分2分别反复经过正己烷:氯仿∶甲醇=2∶1∶1、氯仿∶甲醇=1∶1以及纯甲醇体系进行sephadexlh-20柱层析后重结晶得到51.25mg无色针晶。
实施例5过氧麦角甾醇的制备
1、流份制备:同实施例2。
2、过氧麦角甾醇检识:同实施例2。
3、过氧麦角甾醇提取:在步骤2检识结果的基础上,选择含有过氧麦角甾醇的流份以过氧麦角甾醇斑点为主要观察对象通过hptlc-dpph的方法将含有相同斑点流份合并。将含有过氧麦角甾醇的流份107-121、122-123、124-133、134-151、152-155、156-170、171-207以及208-222合并富集得到浸膏40.67g。将样品∶硅胶为1∶1.5的比例制成硅胶干粉,以样品∶硅胶为1∶50的比例进行硅胶柱层析,洗脱液为石油醚∶乙酸乙酯=15∶1-1∶10,将得到的流份通过hptlc-dpph的方法进行目标成分跟踪,合并后得到组分1。将流份74-79、80-84、85-87、88-92、93-106以及组分1分别通过sephadexlh-20柱层析,其洗脱液体系为正己烷∶氯仿∶甲醇=2∶1∶1,将得到的流份进行目标成分跟踪合并得到组分2。组分2分别反复经过正己烷:氯仿∶甲醇=2∶1∶1、氯仿∶甲醇=1∶1以及纯甲醇体系进行sephadexlh-20柱层析后重结晶得到48.96mg无色针晶。
实施例6过氧麦角甾醇的制备
1、流份制备:同实施例3。
2、过氧麦角甾醇检识:同实施例3。
3、过氧麦角甾醇提取:在步骤2检识结果的基础上,选择含有过氧麦角甾醇的流份以过氧麦角甾醇斑点为主要观察对象通过hptlc-dpph的方法将含有相同斑点流份合并。将含有过氧麦角甾醇的流份107-121、122-123、124-133、134-151、152-155、156-170、171-207以及208-222合并富集得到浸膏39.37g。将样品∶硅胶为1∶1.5的比例制成硅胶干粉,以样品∶硅胶为1∶50的比例进行硅胶柱层析,洗脱液为石油醚∶乙酸乙酯=15∶1-1∶10,将得到的流份通过hptlc-dpph的方法进行目标成分跟踪,合并后得到组分1。将流份74-79、80-84、85-87、88-92、93-106以及组分1分别通过sephadexlh-20柱层析,其洗脱液体系为正己烷∶氯仿∶甲醇=2∶1∶1,将得到的流份进行目标成分跟踪合并得到组分2。组分2分别反复经过正己烷:氯仿∶甲醇=2∶1∶1、氯仿∶甲醇=1∶1以及纯甲醇体系进行sephadexlh-20柱层析后重结晶得到48.61mg无色针晶。
实施例7过氧麦角甾醇的抗肿瘤活性
以a549肺癌细胞、eca-109食道癌细胞以及sgc-7901胃癌细胞为试验对象,研究过氧麦角甾醇的抗肿瘤活性。
1、体外细胞毒性
将过氧麦角甾醇溶于适量dmso,稀释配制终浓度梯度为25、30、40、50、60、80μmol/l的过氧麦角甾醇溶液作用于sgc-7901、a549、eca-109癌细胞,通过mtt法测得不同浓度下不同细胞的生长抑制率情况如表2所示,观察表2可知过氧麦角甾醇对sgc-7901与a549癌细胞的生长抑制效果较好,对于癌细胞的生长抑制率随着浓度的增长快速增加,可以有效的杀伤sgc-7901与a549癌细胞。通过在spss绘制药物浓度与细胞生长抑制率关系曲线图,通过软件分析得出药物作用于sgc-7901、a549、eca-109癌细胞的ic50分别为41.60±0.77、42.22±0.80、74.50±0.35μmol/l。
表2mtt法测定药物对各细胞的生长抑制率
2、ao/eb双荧光染色
选择ao/eb双荧光染色法分别将空白组与不同浓度给药组的sgc-7901细胞进行染色,从图7可见空白组的sgc-7901呈现均匀的绿色荧光且细胞核仍处于自然大小的正常结构。20μmol/l的给药组细胞出现了细胞核固缩,荧光分布不均一,这是细胞出现凋亡的特征,而40μmol/l的给药组细胞在荧光下出现的细胞核固缩程度更高,说明其细胞凋亡程度更强。两个给药组均未出现红色荧光,说明eb并未进入细胞,给药组细胞为早期凋亡细胞。
3、检测细胞凋亡率及其周期
分别使用剂量为20、40、60μmol/l的过氧麦角甾醇溶液作用于sgc-7901癌细胞,用annexinv-fitc与pi进行染色,得到图8。图中q2区域为双阳性染色的晚期凋亡细胞或是坏死细胞,q3为v-fiitc染色早期凋亡细胞,q4为活细胞。从图中可见空白组中晚期凋亡细胞或者坏死细胞占1.63%,早期凋亡细胞为4.29%,而给药20、40、60μmol/l剂量组的晚期凋亡细胞或者坏死细胞占1.83%、1.32%、1.65%,早期凋亡细胞占了总细胞量的5.90%、6.12%、10.4%。由此可见随着药物浓度的增加,早期凋亡细胞的比例也在逐渐增加。
对sgc-7901细胞给予3种不同浓度的药物溶液,分别为20、40、60μmol/l,将其培养24h,使用pi进行染色,经流式细胞仪测得各个浓度下sgc-7901细胞生长周期的变化。从图9中看出空白组的sgc-7901细胞处于g0/g1期为48.38%,处于s期为49.04%,g2/m期为2.58%,而20、40以及60μmol/l的给药组的sgc-7901细胞处于g0/g1期分别为52.17%、57.03%、59.37%,处于s期的分别为45.97%、40.56%、39.44%,处于g2/m期的分别为1.86%、2.42%、1.21%。从中可以看出过氧麦角甾醇可以有效地诱导sgc-7901细胞发生g0/g1期阻滞,从而抑制sgc-7901细胞生长增殖,其阻滞g0/g1期的能力与给药浓度成正相关。
4、活性氧的定性与定量
使用dcfh-da作为染色剂,dcfh-da进入sgc-7901癌细胞后被胞内酯酶水解成为dcfh,dcfh可与过氧麦角甾醇作用于sgc-7901癌细胞产生的活性氧反应生成具有绿色荧光的dcf,在荧光显微镜下观察结果如图10。从图中可见空白组的绿色荧光很淡,说明该组sgc-7901癌细胞所含活性氧较少。rosup可以有效地诱导sgc-7901癌细胞产生活性氧,从而使得图10b的绿色荧光较强。而20、40、60μmol/l的给药组的sgc-7901细胞的绿色荧光强度随着浓度的增加越来越强,其中40μmol/l与60μmol/l剂量组的sgc-7901细胞的荧光较阳性组强。实验表明过氧麦角甾醇可以有效地诱导sgc-7901细胞产生活性氧,这可能是由于过氧键与sgc-7901细胞的亚铁离子发生反应,从而生成活性氧。单纯靠在荧光显微镜观察细胞散发出绿色荧光强度无法客观反映过氧麦角甾醇浓度变化对sgc-7901胃癌细胞内活性氧含量的影响,需要通过流式细胞仪对各组的sgc-7901细胞所产生的活性氧含量进行测定。从图11可以得出通过流式细胞仪对各组细胞荧光度值进行检测,空白组、阳性组、20μmol/l剂量组、40μmol/l剂量组以及60μmol/l剂量组荧光强度分别为5.32×105、8.48×105、6.84×105、1.614×106、3.676×106。即sgc-7901细胞的活性氧含量随着过氧麦角甾醇的浓度增加而增加,当过氧麦角甾醇浓度大于40μmol/l时,细胞内活性氧的含量比阳性药大。
5、线粒体膜电位的定性与定量
线粒体膜电位的下降是早期细胞凋亡的标志。由于线粒体膜电位高低不同时,jc-1在线粒体基质的存在形式不同,从而在荧光显微镜下呈现红色荧光或者绿色荧光,因此使用jc-1染色可以直观反映出线粒体膜电位变化情况。从图12可以看出空白组sgc-7901癌细胞观察不到绿色荧光,说明空白组sgc-7901癌细胞不出现线粒体膜电位的下降,没出现细胞凋亡。随着过氧麦角甾醇浓度的增加,sgc-7901癌细胞红色荧光强度逐渐减弱,而绿色荧光强度逐渐地增强,说明随着过氧麦角甾醇浓度的增加,线粒体膜电位呈下降趋势。通过流式细胞仪测定各组sgc-7901胃癌细胞的红色荧光强度与绿色荧光强度的比值,从而客观的反映出各组线粒体膜电位变化情况。空白组、cccp阳性对照组、20μmol/l剂量组、40μmol/l剂量组以及60μmol/l剂量组的红色荧光强度与绿色荧光强度比值分别为2.88、1.77、2.63、1.9、1.68。从中可以看出随着过氧麦角甾醇浓度的增加,二者的比值急剧下降,当过氧麦角甾醇浓度大于60μmol/l时,其比值比阳性组的小。这说明过氧麦角甾醇可以有效地降低sgc-7901胃癌细胞的线粒体膜电位,呈现剂量依赖性。综合上述结果,表明过氧麦角甾醇可能是通过激活ros介导的线粒体凋亡通路,从而抑制sgc-7901胃癌细胞生长繁殖。
实施例8过氧麦角甾醇的降血糖活性
通过高浓度胰岛素刺激hepg2肝癌细胞,使其产生胰岛素抵抗,从而得到胰岛素抵抗模型。采用酶标仪测定各实验组溶液的葡萄糖含量,比较各组细胞的葡萄糖摄取能力。
从表3可以看出阳性药二甲双胍具有良好的降糖效果,当过氧麦角甾醇给药浓度分别为3.125、6.25、12.5、25、50、100μmol/l时葡萄糖消耗量为6.08±0.59、8.83±1.04、9.64±0.63、10.21±0.86、10.36±0.37、10.39±0.50mmol/l。当剂量为100μmol/l时,二甲双胍组细胞对葡萄糖最大的摄取能力是模型组细胞的1.38倍。而不同浓度给药组的细胞对葡萄糖的消耗量分别为7.30±0.45、7.54±0.51、7.27±1.17、6.70±0.99、8.46±0.56以及9.02±0.38mmol/l。当剂量为100μmol/l时,给药组细胞对葡萄糖最大的摄取能力是模型组细胞的1.20倍,说明过氧麦角甾醇具有较好地促进胰岛素抵抗模型细胞摄取培养液中葡萄糖的能力。通过分子对接的方式将过氧麦角甾醇与14个治疗糖尿病靶点蛋白进行结合能力模拟运算。实验发现过氧麦角甾醇与inos结合自由能为-12.43kcal/mol,inos可能是过氧麦角甾醇改善胰岛素抵抗潜在的作用靶点,其主要结合位点为过氧麦角甾醇的过氧桥与羟基。
从mtt实验结果中得出过氧麦角甾醇对于hepg2肝癌细胞毒性较弱,ic50>100μmol/l。
表3给药组以及阳性组细胞消耗葡萄糖情况
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!