A型塞内卡病毒的微滴数字PCR检测引物、探针及其应用的制作方法
本发明涉及分子生物学
技术领域:
,具体地说,涉及一种a型塞内卡病毒的微滴数字pcr检测引物、探针及其应用。
背景技术:
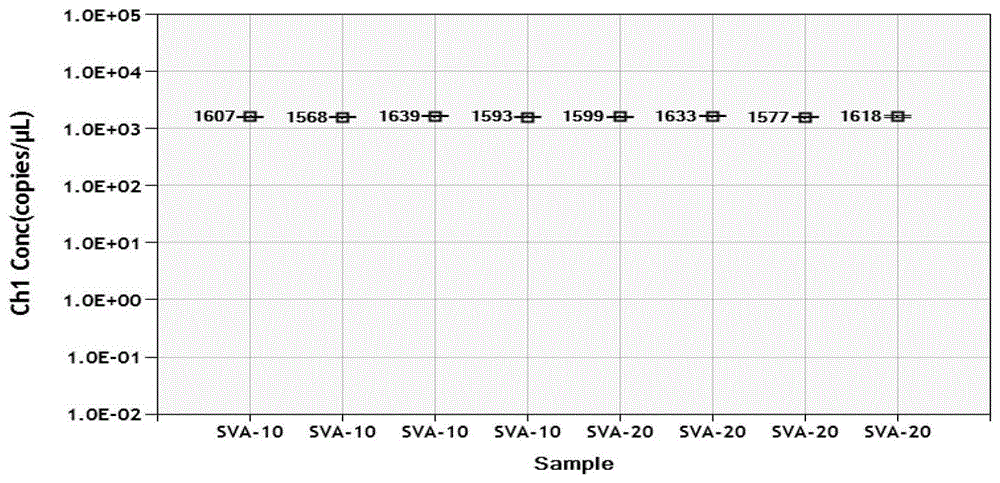
:猪塞内卡病毒病是由a型塞内卡病毒(senecavirusa,sva)引起的一种猪的传染性疾病,可引起仔猪、育肥猪、经产母猪和公猪鼻、唇部、蹄部、腹部、背部等部位出现水泡、脓包、溃疡等,并伴有跛行、站立困难,病猪死亡率可达30%-70%,并且引起发病猪生产性能大幅下降。目前,其表现的临床症状、危害程度等呈现愈来愈严重的趋势,已给养猪业带来了较大的经济损失,因此在临床上需要一种能够快速诊断sva的检测方法。传统的sva检测方法主要包括病毒的分离与鉴定、间接免疫荧光试验(ifa)和间接酶联免疫吸附试验(间接elisa)等。目前最常用核酸检测方法有反转录-聚合酶链反应试验(rt-pcr)和荧光rt-pcr(realtimert-pcr)试验。传统的sva检测方法存在需要使用高成本仪器设备,试验所需时间较长等缺点。rt-pcr和realtimert-pcr虽然较之以往方法具有特异性更强、灵敏度高、重复性好,且自动化程度高等特点,但是上述方法均只能实现定性和半定量的检测,无法对病毒核酸进行精确定量,在灵敏度和敏感性特异性上仍存在一定的局限性。微滴式数字化pcr(dropletdigitalpcr,ddpcr)将传统定量pcr的一个test变成20,000-30,000个test,大大提高了核酸序列检测的灵敏度和精准度。ddpcr系统包括:微滴发生器和微滴分析仪,及其相关的耗材。微滴发生器将每个样品分成20,000-30,000个均匀的纳升级微滴,其中每个微滴或不含待检核酸靶分子,或者含有一个至数个待检核酸靶分子。每个微滴都作为一个独立的pcr反应器。随后微滴在pcr板上,开展终点pcr扩增。采用微滴分析仪(dropletreader)逐个对每个微滴进行检测,有荧光信号的微滴判读为1,没有荧光信号的微滴判读为0,最终根据泊松分布原理以及阳性微滴的比例,分析软件可计算给出待检靶分子的浓度或拷贝数。与传统的定量pcr相比,数字pcr的精确度和灵敏度更佳。利用微滴式数字pcr技术,研究人员可以检测稀有突变,精确测定拷贝数变异,并对基因表达进行绝对定量。目前针对a型塞内卡病毒尚没有理想的微滴数字pcr检测方法,因此需要提供一种a型塞内卡病毒微滴数字pcr检测引物、探针及其应用以解决现有技术的问题。技术实现要素:为了解决现有技术中存在的问题,本发明的目的是提供一种精确度、灵敏度佳的a型塞内卡病毒微滴数字pcr检测引物、探针及其应用。为了实现本发明的目的,本发明的技术方案如下:一种a型塞内卡病毒的微滴数字pcr检测引物,所述引物的核苷酸序列为:上游引物f:5’-tgcaccccttcgctgactacggt-3’(seqidno.1);下游引物r:5’-gagttctcccagaatcgccg-3’(seqidno.2)。一种a型塞内卡病毒的微滴数字pcr检测探针,所述探针的核苷酸序列为:探针p:5’-gccttgttcgactgacctgg-3’(seqidno.3)。优选地,本发明的探针p为:5’-fam-gccttgttcgactgacctgg-mgb-3’该探针可配合本发明的引物使用以进行a型塞内卡病毒的微滴数字pcr检测,因此本发明还提供一种含有上述引物和探针的组合物。本发明引物、探针设计是以genbank已发表的猪塞内卡病毒基因组序列为靶基因,针对sva基因保守区域进行适用于ddpcr的特异性引物及相应的探针设计。此外,本发明还提供一种上述引物在制备a型塞内卡病毒检测试剂或试剂盒中的应用、上述组合物在制备a型塞内卡病毒检测试剂或试剂盒中的应用和含有上述引物或组合物的试剂或试剂盒。本发明的所述试剂盒还包括:微滴发生油、质控液、阳性对照、阴性对照、rna阳性模板、缓冲液中的一种或多种。本发明另提供一种非疾病诊断目的的检测a型塞内卡病毒的方法,该方法包括:(1)提取待测样品的rna,获得微滴数字pcr反应模板;(2)利用上述引物与探针进行微滴数字pcr扩增。本发明方法步骤(2)中,所述微滴数字pcr扩增的反应体系为:one-stepddpcrsupermix5.0μl,reversetranscriptase2.0μl,300mmdtt1.0μl,10μm上游引物1.8μl,10μm下游引物1.8μl,10μm探针0.5μl,水5.9μl,rna模板2.0μl。本发明方法步骤(2)中,所述微滴数字pcr扩增的反应程序为:50℃20min;95℃,10min;94℃30秒,退火温度55℃45秒,40个循环;98℃10min;12℃60min;升降温速率2.5℃/s。上述反应体系及条件适用于从猪组织、血清,或细胞培养液中提取得到的rna样品,可实现对a型塞内卡病毒的精准检测。本发明的有益效果至少在于:采用本发明的引物和探针进行a型塞内卡病毒微滴数字pcr检测特异性好、灵敏度高(可达到1个拷贝)、重复性好,可直接定量,检测方便快捷、结果准确可靠。附图说明图1为本发明实施例2的反转录时间为10min(sva-10)、20min(sva-20)条件下数字pcr实验结果图;图2为本发明实施例2的退火温度的优化实验结果图,其中,a01,a02,a03,a04分别对应温度为54℃,55℃,58℃,60℃的实验结果;图3为本发明实施例2的升降温速度的优化实验结果图,其中,图3a的升降温速度为1℃/s;图3b的升降温速度为2.5℃/s;图3c的升降温速度为5℃/s;图4为本发明实施例3的数字pcr梯度稀释扩增结果图,其中,a01、b01、c01代表sva的标准物质梯度稀释为10-1时的结果,e01、f01、g01代表sva的标准物质梯度稀释为10-2时的结果,a02、b02、c02代表sva的标准物质梯度稀释为10-3时的结果,e02、f02、g02代表sva的标准物质梯度稀释为10-4时的结果;图5为本发明实施例3的荧光定量pcr梯度稀释检测结果;其中,图中自左至右1-4分别对应的稀释度为10-1,10-2,10-3,10-4;图6为本发明实施例3的荧光定量pcr结果通过梯度稀释建立的线性图谱;图7为本发明实施例7的临床样品部分测试结果图,其中,b02代表一份临床阳性样品,f03、g03、h03分别代表一份临床阴性样品。具体实施方式下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。本发明实施例中所用主要原材料来源见表1。表1其中,猪塞内卡病毒株v202013于2020年1月14日保藏于中国典型培养物保藏中心(简称cctcc,地址:中国武汉武汉大学,中国典型培养物保藏中心,邮编430072),保藏编号为cctccno:v202013,分类命名为:塞内卡病毒svayrq/2016。实施例1引物及探针的设计与筛选本实施例根据genbank已发表的猪塞内卡病毒基因组序列,针对sva基因保守区域进行适用于ddpcr的特异性引物及相应的探针设计,获得了多种引物及探针的组合方式,在对各组合方式进行特异性、灵敏度、重复性等多方面筛选后,获得了最优引物及探针序列如下:上游引物f:5’-tgcaccccttcgctgactacggt-3’(seqidno.1);下游引物r:5’-gagttctcccagaatcgccg-3’(seqidno.2)。探针p:5’-fam-gccttgttcgactgacctgg-mgb-3’(seqidno.3)。具体设计、筛选过程如下:1、本发明引物、探针设计:根据genbank已发表的猪塞内卡病病毒3d基因组序列为靶基因,进行适用于ddpcr的特异性引物和探针的设计。本实施例给出了筛选最佳引物的过程,选择用软件设计出的其中几对备选引物、探针进行筛选,备选引物、探针如下,见表2。共设计4对引物、探针。表22、引物的筛选(1)引物随机配为四对:frp、f2r2p2、f3r3p3、f4r4p4。(2)然后采用4对引物及探针针对sva标准物质(国家二级标准物质gbw(e)091053)相同浓度的核酸进行数字pcr检测。(3)检测过程具体为:在ddpcr平台上进行检测,ddpcr体系:2xone-steprt-ddpcrsupermix10μl,上游引物,下游引物分别为1μl,探针0.5μl,rnasefreeddh2o3.5μl,阳性模板4μl,总体积20μl(引物和探针的浓度均为10μm)。然后生成微滴。pcr仪扩增,为50℃反转录10min;95℃预变性10min;94℃变性30sec,60℃退火60sec,共40个循环;98℃10min结束反应。微滴数字pcr检测仪检测。(4)检测结果参见表3。表3编号检测结果(copies/μl)frp1.68e+04f2r2p21.15e+04f3r3p31.13e+04f4r4p41.22e+04(5)分析结果:根据实验结果选择fr+p引物探针组合。实施例2数字pcr反应体系及程序的筛选本实施例所用的引物、探针指实施例1中所记载的引物、探针。1、数字pcrsupermix的选择反转录酶选择amv、superscripttmiii酶。以sva病毒rna为模板,用2×ddpcrsupermixforprobes体系加入amv、superscripttmiii一步法反转录酶进行数字pcr。第一组:用superscripttmiii酶:2×ddpcrsupermixforprobes10μl,superscripttmiii1μl,10μm引物f、r各1.8μl,10μm探针0.5μl,水4.7μl,模板2.0μl。反应条件:50℃60min;95℃10min;95℃30s,55℃1min,40个循环;98℃10min;4℃60min;升降温速率2℃/s。第二组:用amv酶:2×ddpcrsupermixforprobes10.0μl,amv0.2μl,10μm引物f、r各1.8μl,10μm探针0.5μl,水5.5μl,模板2.0μl。反应条件:42℃60min;95℃10min;95℃30s,55℃1min,40个循环;98℃10min;4℃60min;升降温速率2℃/s。第三组:用one-steprt-ddpcrkitforprobes试剂盒:one-stepddpcrsupermix5.0μl,reversetranscriptase(酶)2.0μl,300mmdtt1.0μl,10μm引物f、r各1.8μl,10μm探针0.5μl,水7.7μl,模板2.0μl。反应条件:42℃60min;95℃10min;95℃30s,55℃1min,40个循环;98℃10min;4℃60min;升降温速率2℃/s。结果:用superscripttmiii酶做的结果微滴平均生成数目为7474;用amv酶做的结果微滴平均生成数目为7737;用one-steprt-ddpcrkitforprobes试剂盒做的结果,微滴平均生成数目为12670。实验证明,用其他体系的酶反应实验不能与伯乐的微滴生成油匹配,所以微滴生成数减少。最后选择用one-steprt-ddpcrkitforprobes试剂盒为后期实验试剂盒。2、引物探针浓度的优化影响数字pcr检测结果的关键因素之一是引物探针的浓度,浓度过低会导致拷贝数降低,浓度过高会导致pcr反应抑制。本实施例利用病毒灭活液用磁珠法自动提取试剂,提取病毒sva核酸标准物质为模板,ddpcr实验反应体系为:one-stepddpcrsupermix5.0μl,reversetranscriptase(酶)2.0μl,300mmdtt1.0μl,10μm引物f、r各1.8μl/1.5μl/2.0μl,10μm探针0.5μl/0.3μl/0.6μl,模板2.0μl,用水补齐至20μl。反应条件:50℃20min;95℃10min;94℃30s,55℃1min,40个循环;98℃10min;4℃60min;升降温速率2.5℃/s。引物设置了600,900,1200nmol/l的浓度,探针相应设置了150,250,350nmol/l三种浓度。测试结果见表4。表4引物探针浓度优化的结果分析其中,第一组:引物浓度600nm,探针浓度150nm;第二组:引物浓度900nm,探针浓度250nm;第三组:引物浓度1200nm,探针浓度350nm。三组的rsd值均小于5%,但是第一组,第三组,拷贝数明显低于第二组,因此,引物浓度定为第二组900nm,探针浓度250nm。实验结果及分析:设置反应条件,引物探针浓度为900/250nmol/l时,拷贝值最高,数值最稳定。结论:900nmol/l的引物和250nmol/l的探针反应浓度为最佳引物探针浓度。3、反转录实验sva的标准物质是rna样本,在pcr定值时需要把rna反转录为cdna后再进行后续的实验,在一步法pcr的试剂里含有两种反应酶液,一种是反转录酶,功能是把rna转化为cdna,另一种是dna聚合酶,用于后续的pcr扩增。在进行反转的时候,dna聚合酶是完全没有活性的。该实验目的是验证反转录不同时长对最后结果的影响。(1)用同一份核酸标准物质提取一份rna,作为此次实验的模板。(2)按one-stepddpcrsupermix5.0μl,reversetranscriptase(酶)2.0μl,300mmdtt1.0μl,10μm引物f、r各1.5μl,10μm探针0.5μl,水8μl,模板2.0μl。配置10份ddpcr实验试剂。(3)生成8份ddpcr微滴。(4)分出4份pcr扩增程序是50℃,10min;95℃,10min;94℃,30秒,退火温度55℃60秒,40个循环;98℃,10min;升降温速率2.5℃/s的程序,用1号pcr仪进行检测。(5)分出4份pcr扩增程序是50℃,20min;95℃,10min;94℃,30秒,退火温度55℃60秒,40个循环;98℃,10min;升降温速率2.5℃/s的程序,用2号pcr仪进行检测。注:1、2号分别用的是两台同品牌同类型的pcr仪。(6)实验结果见图1,其中sva-10代表反转录10分钟,sva-20代表反转录20分钟。本实验采用两种反转录时间验证反转录的时长对数字pcr结果的影响,结果拷贝数差异不大。sva反转录10分钟,与反转录20分钟的结果配对t检验的结果(见表5)均大于0.05,显示是无显著性差异。表510min、20min反转录时长结果t检验表结论:数字pcr实验是一步法pcr实验,在反转录10分钟与反转录20分钟结果差异不大,最后选择20分钟的反转录时间。4、退火温度优化退火温度是数字pcr实验中很关键的影响因素,它过低会影响引物和探针与模板的结合效率,温度过高会影响微滴的扩增效率。本实施例退火温度摸索了54℃,55℃,58℃,60℃四个温度,跨度是6度;反应体系为:supermix5.0μl,reversetranscriptase2.0μl,300mmdtt1μl,10μm引物(f)1.8μl,10μm引物(r)1.8μl,10μm探针0.5μl,水5.9μl,模板2.0μl。反应条件为:50℃20min;95℃10min;94℃30s,54、55、58、60℃60s,40个循环;98℃10min;12℃60min。升降温速率2.5℃/s。实验结果参见图2,退火温度在60℃时,信号强度有所下降,拷贝数降低。因此,将退火温度定为55℃。结论:pcr扩增程序是50℃20min;95℃10min;94℃30秒,退火温度55℃60秒,40个循环;98℃10min;12℃60min;升降温速率2.5℃/s。退火温度为55℃为最佳反应条件。5、升降温速度的优化升降温速度是数字pcr实验中很关键的影响因素,升降温速度过快会影响微滴的稳定性,也会影响数字pcr扩增效率,退火温度过快,或过慢均会影响实验效果,导致阳性微滴散乱。通过升降温速度的优化实验,以找到最优的升降温速度。以优化sva数字pcr检测方法,实验配制体系为supermix5.0μl,reversetranscriptase2.0μl,300mmdtt1μl,10μm引物(f)1.8μl,10μm引物(r)1.8μl,10μm探针0.5μl,水5.9μl,模板2.0μl。pcr扩增程序是50℃20min;95℃10min;94℃30秒,55℃60秒,40个循环;98℃10min;12℃60min;升降温速度设置了1℃/s,2.5℃/s,5℃/s。进行优化。结果:1℃/s,2.5℃/s阳性微滴和阴性微滴分布集中,5℃/s阳性微滴和阴性微滴分布有些扩散。在1℃/s,2.5℃/s扩增效果接近的情况下,选择2.5℃/s的升降温速度,以节约实验时间。结果如图3所示。其中,图3a的升降温速度为1℃/s;图3b的升降温速度为2.5℃/s;图3c的升降温速度为5℃/s。经上述优化实验后,本实施例最终确定的sva数字pcr检测体系和程序为:sva数字pcr检测方法,实验配制体系为:supermix5.0μl,reversetranscriptase2.0μl,300mmdtt1μl,10μm引物(f)1.8μl,10μm引物(r)1.8μl,10μm探针0.5μl,水5.9μl,模板2.0μl。pcr扩增程序为:50℃20min;95℃10min;94℃30秒,退火温度55℃45秒,40个循环;98℃10min;12℃60min;升降温速率2.5℃/s。实施例3svaddpcr检测方法的动态范围及灵敏度测定将10倍倍比稀释的已制备好的含有sva的标准物质梯度稀释10-1,10-2,10-3,10-4,每个浓度做3个重复检测,同时在微滴式数字pcr检测和荧光定量pcr平台实验,测试结果见表6、图4(数字pcr结果),图5至图6。数字pcr体系one-stepddpcrsupermix5.0μl,reversetranscriptase(酶)2.0μl,300mmdtt1.0μl,10μm引物(f)1.8μl,10μm引物(r)1.8μl,10μm探针0.5μl,水5.9μl,模板2.0μl。反应程序:50℃20min;95℃10min;94℃30秒,退火温度55℃45秒,40个循环;98℃10min;12℃60min;升降温速率2.5℃/s。数字pcr结果分析:将10倍倍比稀释的已制备好的含有sva的标准物质梯度稀释10-1,10-2,10-3,10-4,每个浓度做3个重复检测,当检测浓度低到100拷贝以下时能稳定检出且cv值为1.8%小于25%,最终确定该方法最低检测下限为3.2拷贝/μl。数字pcr结果通过梯度稀释建立线性图谱,计算出r2=1>0.99,线性关系良好,线性动态范围灵敏度为3.2×100-2.5×103拷贝/μl(copies/μl),最低线性范围内检测下限为3.2×100拷贝/μl。表6倍比稀释(10倍)样本检测结果sva荧光pcr检测方法,实验配制体系为:qpcrsupermix,10μl,模板2μl,引物探针混和液4μl,用水补齐至20μl配制体系。qpcr扩增程序为:50℃20min;95℃,10min;94℃30秒,退火温度55℃60秒,40个循环;98℃10min;12℃60min;升降温速率2.5℃/s,行荧光定量扩增实验。荧光定量pcr梯度稀释检测结果参见图5(检测sva敏感性扩增曲线)。荧光定量pcr结果通过梯度稀释建立的线性图谱参见图6。荧光定量pcr结果分析:荧光定量pcr扩增的标准曲线为:y=-3.289x+37.497,标准曲线的r2值为0.999,扩增效率为101.4%。结论:数字pcr的的检测线性动态范围灵敏度为3.2×100-2.5×103copies/μl,最低线性检测下限为3.2×100copies/μl。荧光定量pcr的最低检测下限为31(ct值)。实施例4svaddpcr检测1、病毒rna提取模板rna的制备:以sva毒株为阳性对照,bhk21细胞液为阴性对照,同时采用trizol法提取总rna。具体操作如下:分别取sva毒株、阴性对照及待测样品各200μl于1.5ml离心管中,再加入600μl的trizol于涡旋器震荡2-3min,加入200μl氯仿,离心后,取上清转入另1.5ml离心管中,加200μl异丙醇沉淀,75%乙醇洗涤沉淀,干燥,最后用20μldepc(焦磷酸二乙酯)水溶解沉淀,取10μl用于反转录,其余-20℃保存。2、微滴数字pcr绝对定量检测试剂盒的组装将试剂盒分为一步法微滴数字pcr检测试剂,方便保存和运输。用合适的外包装盒包装以下试剂,贴标签(标示名称、批号、生产日期、有效期等)。溶液a(rnasefreeddh2o)1支,1ml;溶液b(一步法ddpcr探针法预混液)一支,900μl;溶液c(探针法ddpcr微滴发生油)一支,7ml;溶液d为ddpcr探针法质控液,3.5ml;溶液e(阳性对照)一支,40μl;溶液f(阴性对照)一支,40μl;使用基因组rna为阳性模板,使用tris-edta缓冲液(0.01mph8.0)稀释后冷冻保存。将检验合格的阳性对照制剂按650μl定量分装。使用bhk21细胞液为阴性对照。上述一步法ddpcr探针法预混液其900μl的配方为:500μl的2xone-steprt-ddpcrsupermixforprobes,实施例1中的上、下游引物分别为90μl,探针为40μl,引物和探针的浓度均为10μm。rnasefreeddh2o180μl。使用时,在18μl的ddpcr探针法预混液中加入2μl的rna模板,得到20μl的ddpcr反应液,用于制备微滴。3、ddpcr方法的建立(1)制备ddpcr反应液,总体积20μl。向pcr扩增管中加入下列反应物,见表7。表7单反应体系配方一步法ddpcr探针法预混液18μl模板rna2μl空白对照:以bhk21细胞液代替模板,同样条件下扩增。(2)取微滴发生卡置于卡托中固定,将20μl反应液加入到微滴发生卡中间一排的8个孔内,不足8个样品时用20μl1×溶液d补足,使用8通道20μl移液器和20μl滴头(不能用200ul滴头),加样时枪头接近孔一侧底部,与侧壁呈大约15°角,缓慢打出液体,打出一部分后慢慢提升移液头位置再打出余下液体,不要将移液器按至超过第一档位置以免引入气泡。(3)在微滴发生卡最底下一排8个孔中各加入70μl微滴生成油,同样不能有空着的孔。(4)盖上胶垫,两边的小孔都要钩牢。(5)将以上卡托轻轻地平稳放置于微滴生成仪中,开始生成微滴,注意仪器上指示灯状态,一般2分钟之内完成。(6)微滴生成于微滴发生卡最上面一排孔内,建议使用8通道移液器和200μl滴头小心缓慢吸取,调整吸取体积为40μl,将卡托放平,枪头以与孔壁呈30~45°角放入,轻触孔底,约5秒吸取40ul,再同样缓慢地打入96孔板相应位置孔内(约5秒),滴头贴近孔壁接近孔底,注意封上盖以防油挥发,每次弃去已使用过的微滴发生卡和胶垫。(7)转移油滴进入96孔板内完成后,用预热好的px1热封仪对其进行封膜(膜亮面朝上,暗面向下),推荐的运行程序为:180℃,10s,一般无需颠倒方向二次封膜;封好膜之后应该在30分钟内进行pcr反应,或者放于4℃冰箱4小时之内进行pcr(可在任意一台96孔pcr仪上完成),升降温速度≤2.5℃/s。反应条件见表8。表8(8)将之前完成pcr的96孔板放入plateholder中组装好,注意板斜角方位,组装好之后轻轻地平稳放入微滴读取仪中。(9)打开quantasoft软件,每次实验之前做一次系统清洗(若一周以上未使用,先做一次填充油路再做系统清洗)。之后对96孔板中样品信息进行设置,提供实验名称、实验类型以及探针信息等,完成后即可运行仪器,结束后结果会被自动分析,人工核实后保存结果。4、结果分析和判定本发明试剂盒检测结果的判定方法为:(1)阳性对照:20±2个拷贝;(2)阴性对照:<1个拷贝;待测样本结果判定:(1)阳性:标本检测结果≥1个拷贝。(2)阴性:标本检测结果<1拷贝。实施例5特异性检验用实施例4中制备的试剂盒以实施例4中的方法,分别检测猪塞内卡病毒a、口蹄疫病毒、猪繁殖与呼吸综合征病毒、猪瘟病毒、猪圆环病毒2型、猪流行性腹泻病毒、猪传染性胃肠炎病毒,结果全为阴性;检测sva的组织与细胞培养物,结果全为阳性,具体检测对象及结果见表9。表9以上检测结果证明了本发明的试剂盒特异性好、灵敏度高、可直接定量,检测方便快捷、结果准确可靠。实施例6重复性试验在同一份样品中,重复取样10次,每次2μl,使用实施例4的数字pcr方法检测。最后计算变异系数为2.39%。结果表明,本发明的数字pcr方法重复性好,检测结果稳定、可靠。具体结果见表10。表10实施例7临床样品验证1、临床样品检测对河南省动物疫病预防控制中心收集保存的40份猪淋巴结、心脏、肺脏、脾脏、肠道、气管、肾脏等组织(其中sva阳性样品9份)进行检测。2、实验方法对收集保存的40份猪组织(其中sva阳性样品9份)研磨后,使用全自动核酸提取仪进行rna提取,并用本发明建立的rt-ddpcr方法(实施例4的方法)进行检测。有阳性微滴即可判定为sva阳性,分析本方法临床样品检测效果。3、实验结果40份临床样品中,9份sva阳性样品全部检测为阳性,其余样品检测为阴性。部分检测结果见图7。4、实验结论:该rt-ddpcr方法检测结果与sva基因测序结果符合率为100%。虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。序列表<110>河南省动物疫病预防控制中心<120>a型塞内卡病毒的微滴数字pcr检测引物、探针及其应用<130>khp191117179.7<160>12<170>siposequencelisting1.0<210>1<211>23<212>dna<213>人工序列(artificialsequence)<400>1tgcaccccttcgctgactacggt23<210>2<211>20<212>dna<213>人工序列(artificialsequence)<400>2gagttctcccagaatcgccg20<210>3<211>20<212>dna<213>人工序列(artificialsequence)<400>3gccttgttcgactgacctgg20<210>4<211>21<212>dna<213>人工序列(artificialsequence)<400>4cctctctttccgacgctgttt21<210>5<211>22<212>dna<213>人工序列(artificialsequence)<400>5tataagccgtcgttgttttgga22<210>6<211>16<212>dna<213>人工序列(artificialsequence)<400>6tctaaaacgcaaattc16<210>7<211>20<212>dna<213>人工序列(artificialsequence)<400>7cctctctttccgacgctgtt20<210>8<211>21<212>dna<213>人工序列(artificialsequence)<400>8ataagccgtcgttgttttgga21<210>9<211>15<212>dna<213>人工序列(artificialsequence)<400>9ctaaaacgcaaattc15<210>10<211>22<212>dna<213>人工序列(artificialsequence)<400>10ctctttccgacgctgtttttct22<210>11<211>23<212>dna<213>人工序列(artificialsequence)<400>11tgtataagccgtcgttgttttgg23<210>12<211>13<212>dna<213>人工序列(artificialsequence)<400>12aaacgcaaattcg13当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1