一种抗菌药物及其制备方法与流程
本发明属于化学合成领域,特别是涉及一种抗菌药物及其制备方法。
背景技术:
氟康唑(fcz)作为被用于全身性真菌感染的抗真菌三唑类药物,具有低毒、易于吸收、生物利用度高、作用范围大等优点。可是由于氟康唑的滥用,使真菌对其的耐药性愈发严重,所以在对氟康唑的深入改造方面就至关重要。
四唑环作为富电子五元平面共轭体系,具备电子给受体的双重特性。这一特性使其能够形成多种的次级键,如氢键、金属配位、π-π堆积、静电作用,这样可以表现出更多不同的性能,使四唑类化合物被广泛应用于众多领域。其中医药方面,一些四唑类化合物本身表现出广泛的生物活性,包括抗菌性能和抗真菌性能,并且已经取得了优秀的临床数据[见muszalskai,j.pharm.sci.,2014,103(1):2-28];同时,在药物设计与开发过程中,四唑环能够被用来改善已知药物的生物活性,同时四唑环的引入往往可以增强药物的功效及延长药物的作用时间[见v.a.ostrovskii,russianchemicalbulletin,2012,61(4):768-780]。除此之外,通常不会有伴随急性毒性的增加[见v.a.ostrovskii,comp.heterocycl.chem.iii,2008,6:257]。四唑环是现代药物分子设计中重要的结构片段,它可以通过多种相互作用与生物体内多种酶或受体等作用靶点结合,表现出优异的生物活性。
基于以上背景,在保留氟康唑上大部分具有药效的结构基础上,拟对氟康唑进行修饰改造。
技术实现要素:
本发明的目的在于提供一种通过引入四唑环而具有更广谱的抗微生物活性(相比氟康唑)的抗菌药物及其制备方法。
为了达成上述目的,本发明的技术方案是:
一种抗菌药物,所述抗菌药物为2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇,所述抗菌药物的化学结构式为:
采用上述技术方案后,本发明一种抗菌药物,具有以下有益效果:该化合物在氟康唑基础上进行修饰改造,引入四唑环,相比氟康唑,本发明的化合物具有更广谱的抗微生物活性。
一种所述的抗菌药物的制备方法,包括如下步骤:
(1)以氯乙酰氯、间二氟苯为原料,并以氯化铝为催化剂,室温反应,冰浴下加入盐酸水溶液,析出固体,过滤后所得固体用溶剂溶解,旋蒸除去溶剂得到2-氯-2',4'-二氟苯乙酮;
(2)将1,2,4-1h-三氮唑和步骤(1)得到的2-氯-2',4'-二氟苯乙酮溶解于乙酸乙酯中,滴加三乙胺,反应6~8小时后,过滤,滤液以设定比例的乙酸乙酯和石油醚作为洗脱剂通过层析柱分离,得到α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮;
(3)将步骤(2)中α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮溶解于溶剂中,再加入三甲基碘化亚砜和三甲基十六烷基溴化铵,反应温度为40~80℃,滴加质量浓度20%的氢氧化钠水溶液,反应1~5小时后,经萃取、水洗,旋蒸除去溶剂,残留液用乙酸乙酯稀释,在冰浴条件下再滴加甲烷磺酸的乙酸乙酯溶液,析出固体,所得固体用热乙醇溶解,完全溶解后析出固体,过滤得到1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑;
(4)将1h-四氮唑和步骤(3)得到的1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑溶解于乙醇中,滴加三乙胺,反应温度升至设定温度,反应9~12小时后,以设定比例的乙酸乙酯和石油醚作为洗脱剂通过层析柱分离,得到2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇。
进一步地,步骤(1)中,间二氟苯与氯乙酰氯的摩尔比为1:1~1.2;间二氟苯与氯化铝的摩尔比为1:1~1.2。
进一步地,所述的步骤(1)中,盐酸水溶液中盐酸和水的体积比为1:1-1:5,氯化铝:盐酸:水的质量比为1:1:1-1:3:15。
进一步地,步骤(2)中,2-氯-2',4'-二氟苯乙酮与1,2,4-1h-三氮唑的摩尔比为1:1~1.2;2-氯-2',4'-二氟苯乙酮与三乙胺的摩尔比为1:1~1.2。
进一步地,步骤(2)中,乙酸乙酯与石油醚的体积比为5:1-5:5。
进一步地,所述的步骤(3)中,所用溶剂为甲苯、苯或环己烷。
进一步地,所述的步骤(3)中α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮与三甲基碘化亚砜的摩尔比为1:1;α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮与氢氧化钠的摩尔比为1:3~1:9。
进一步地,步骤(4)中,乙酸乙酯与石油醚的体积比为5:1-5:5。
进一步地,步骤(4)中,1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑与1h-四氮唑的摩尔比为1:1~1.2;1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑与三乙胺的摩尔比为1:1~1.2。
采用上述技术方案后,本发明一种抗菌药物的制备方法,具有以下有益效果:该方法在保留氟康唑上大部分具有药效的结构基础上,通过引入四唑环得到2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇。
附图说明
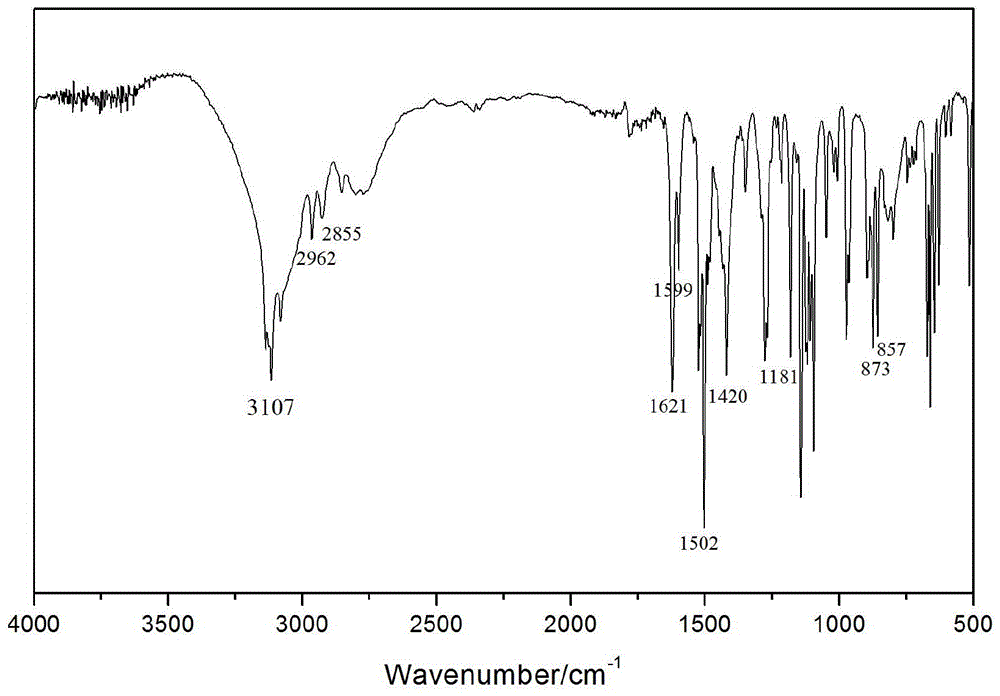
图1是本发明2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇的傅里叶变换红外光谱图。
图2是本发明2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇的核磁共振氢谱图。
图3是本发明2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇的核磁共振碳谱图。
具体实施方式
为了进一步解释本发明的技术方案,下面通过具体实施例来对本发明进行详细阐述。
实施例1
一、制备方法
本发明一种抗菌所述抗菌药物为2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇(tfcz),所述抗菌药物的化学结构式为:
本发明一种如上述的抗菌药物的制备方法,其合成路线如下所示:
本发明一种如上述的抗菌药物的制备方法,包括如下步骤:
(1)2-氯-2',4'-二氟苯乙酮的制备:室温下,依次向已干燥的100ml三口瓶中加入无水氯化铝6.68g和间二氟苯5.71g。在磁力搅拌5min下,用恒压滴液漏斗向三口瓶中缓慢地滴加氯乙酰氯5.65g,3h后反应完毕。冰浴下向三口瓶中加入盐酸水溶液(盐酸和水的体积比为1:2),析出大量固体,过滤干燥得粗产品;粗产品用10ml二氯甲烷溶解,过滤除去不溶性杂质,滤液通过旋转蒸发仪除去溶剂(即二氯甲烷),得到纯的2-氯-2',4'-二氟苯乙酮8.66g,产率91%。
其中,2-氯-2',4'-二氟苯乙酮的合成路线如下:
(2)α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮的制备:依次向带冷凝管的100ml三口瓶中加入2-氯-2',4'-二氟苯乙酮1.91g和1,2,4-1h-三氮唑0.69g,再加乙酸乙酯溶解。待溶解完全后,在磁力搅拌下用恒压滴液漏斗缓慢地滴加三乙胺1.01g,升温至60℃,反应6-8小时,优选地,8h后反应完毕,过滤,滤液浓缩。再以乙酸乙酯:石油醚体积比=5:2(v/v)的洗脱剂,通过层析柱分离,再将分离出来的物质通过红外和核磁检测,发现最后分离出来的产物为最终产物,得到α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮0.74g,产率33%。
其中,α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮的合成路线如下:
(3)1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑的制备:向已干燥的100ml三口瓶中加入α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮2.23g和10ml苯,溶解后加入三甲基碘化亚砜2.20g和相转移催化剂三甲基十六烷基溴化铵0.1g,温度升至40~80℃,优选为60℃,在磁力搅拌下用恒压滴液漏斗缓慢地滴加20%(质量浓度)naoh水溶液(naoh1.2g),通过tlc监测反应进度,反应时间为1-5小时,优选地,3h后反应完毕。分离出甲苯层,水层用甲苯萃取3次,再将甲苯层合并,水洗至中性,通过旋蒸将大部分甲苯旋蒸除去,残留液加乙酸乙酯稀释,在冰浴下滴加甲烷磺酸的乙酸乙酯溶液(甲烷磺酸1g),析出固体,过滤,所得固体用热无水乙醇溶解,待冷却(可置于冰箱中)析出白色固体,过滤干燥得纯的1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑2.16g,产率65%。
其中,1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑的合成路线如下:
(4)2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇的制备:依次向带冷凝管的100ml三口瓶中加入1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑0.50g和1h-四氮唑0.11g,并加10ml乙醇以溶解,再加入1ml三乙胺,升温至60℃回流,通过tlc监测反应进度,反应时间为9~12小时,优选地,9h后反应完毕。再以乙酸乙酯:石油醚=2:1(v/v)的洗脱剂,通过层析柱分离,再将分离出来的物质通过红外(结果如图1所示)和核磁检测(结果如图2和图3所示),发现最后分离出来的产物为最终产物,得到2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇0.13g,产率29%。
其中,2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇的合成路线如下:
需要说明的是,本发明步骤(1)中,间二氟苯与氯乙酰氯的摩尔比为1:1~1.2;间二氟苯与氯化铝的摩尔比为1:1~1.2;盐酸水溶液中盐酸和水的体积比为1:1-1:5,氯化铝:盐酸:水的质量比为1:1:1-1:3:15。
步骤(2)中,2-氯-2',4'-二氟苯乙酮与1,2,4-1h-三氮唑的摩尔比为1:1~1.2;2-氯-2',4'-二氟苯乙酮与三乙胺的摩尔比为1:1~1.2。
步骤(3)中,所用溶剂除了采用上述苯外,还可采用甲苯或环己烷;α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮与三甲基碘化亚砜的摩尔比为1:1;α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮与氢氧化钠(naoh)的摩尔比为1:3~1:9。
步骤(4)中,1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑与1h-四氮唑的摩尔比为1:1~1.2;1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑与三乙胺的摩尔比为1:1~1.2。
二、抗菌性能测试
采用美国临床实验室标准化委员会(nccls)所规定的标准化抗菌敏感性实验方法,在96孔板上利用微量肉汤稀释法对所有目标化合物及参比样的体外抗细菌、抗真菌活性进行测试,并用mic80来表示其抗菌活性,活性数据详见表1。其中,所选测试真菌为白色念珠菌(c.albicans),所选测试细菌包括革兰氏阳性菌金黄色葡萄球菌(s.aureus)和革兰氏阴性菌大肠杆菌(e.coli),并选择氟康唑(fcz)作为参比样。
表1tfcz和fcz的最低抑菌浓度mic80
由上表中可知,相对于fcz,本发明的产物tfcz具有更为广谱性质的抗微生物活性,且对e.coli、s.aureus以及c.albicans均表现出一定的生物活性。其中,tfcz和fcz对c.albicans的mic80分别是4μg/ml和0.25μg/ml。在fcz上引入四唑环后,所得产物tfcz对c.albicans的抗真菌作用相对于fcz变得更弱。可能是因为在fcz上引入四唑环,所合成得到的化合物是手性小分子,且由于化学药物如果含有手性因素,那么该药物的对映体处于生物体内的毒性、药理作用以及生理代谢过程均有着显著的区别,所以可能在得到的产物中存在无效乃至于有害的对映体,导致所合成的化合物对c.albicans的抗菌活性没有起到预期的作用。其中,fcz对e.coli以及s.aureus都没有呈现出抑制作用,而tfcz对e.coli和s.aureus的mic80分别是128μg/ml和16μg/ml。在fcz上引入四唑环后,所得产物tfcz对所测细菌的抗菌活性相较于氟康唑得到了显著的提高,特别是对革兰氏阳性菌s.aureus的抗菌活性。
实施例2
本实施例与实施例1的区别在于步骤(1):本实施例中,2-氯-2',4'-二氟苯乙酮的制备方法如下:室温下,依次向已干燥的100ml三口瓶中加入无水氯化铝8.01g和间二氟苯5.71g。在磁力搅拌5min下,用恒压滴液漏斗向三口瓶中缓慢地滴加氯乙酰氯6.78g,3h后反应完毕。冰浴下向三口瓶中加入盐酸16.02g和水32.04g,析出大量固体,过滤干燥得粗产品;粗产品用10ml二氯甲烷溶解,过滤掉不溶性杂质,滤液通过旋转蒸发仪除去溶剂,得到纯的2-氯-2',4'-二氟苯乙酮8.19g,产率86%。
实施例3
本实施例与实施例1的区别在于步骤(2):本实施例中,α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮的制备方法如下:依次向带冷凝管的100ml三口瓶中加入2-氯-2',4'-二氟苯乙酮1.91g和1,2,4-1h-三氮唑0.83g,再加乙酸乙酯溶解。待溶解完全后,在磁力搅拌下用恒压滴液漏斗缓慢地滴加三乙胺1.21g,升温至回流温度,6h后反应完毕。再以乙酸乙酯:石油醚=5:2的洗脱剂,通过层析柱分离,再将分离出来的物质通过红外和核磁检测,发现最后分离出来的产物为最终产物,得到α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮1.05g,产率47%。
实施例4
本实施例与实施例1的区别在于步骤(3):本实施例中,1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑的制备方法如下:向已干燥的100ml三口瓶中加入α-(1h-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮2.23g和10ml甲苯,溶解后加入三甲基碘化亚砜2.20g和相转移催化剂三甲基十六烷基溴化铵0.1g,温度升至60℃,在磁力搅拌下用恒压滴液漏斗缓慢地滴加20%naoh水溶液(naoh3.6g),通过tlc监测反应进度,5h后反应完毕。分离出甲苯层,水层用甲苯萃取3次,再将甲苯层合并,水洗至中性,通过旋蒸将大部分甲苯旋掉,残留液加乙酸乙酯稀释,在冰浴下滴加甲烷磺酸的乙酸乙酯溶液(甲烷磺酸1g),析出固体,过滤,所得固体用热乙醇溶解,待冷却析出白色固体,过滤干燥得纯的1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑1.40g,产率42%。
实施例5
本实施例与实施例1的区别在于步骤(4):本实施例中,2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇的制备方法如下:依次向带冷凝管的100ml三口瓶中加入1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1h-1,2,4-三氮唑0.50g和1h-四氮唑0.13g,加10ml无水乙醇溶解,再加入1ml三乙胺,加热回流,通过tlc监测反应进度,12h后反应完毕。再以乙酸乙酯:石油醚=2:1(v/v)的洗脱剂,通过层析柱分离,再将分离出来的物质通过红外和核磁检测,发现最后分离出来的产物为最终产物,得到2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)-3-(1h-1,2,3,4-四唑-1-基)-2-丙醇0.23g,产率50%。
上述实施例和附图并非限定本发明的产品和制备方法,任何所属技术领域的普通技术人员对其所做的适当变化或修饰,皆应视为不脱离本发明的专利范畴。
- 还没有人留言评论。精彩留言会获得点赞!