一类N-苄基取代的二脒那秦衍生物及其药物用途的制作方法
本发明属于制药领域,提供一类n-苄基取代的二脒那秦衍生物及其药物用途。
背景技术:
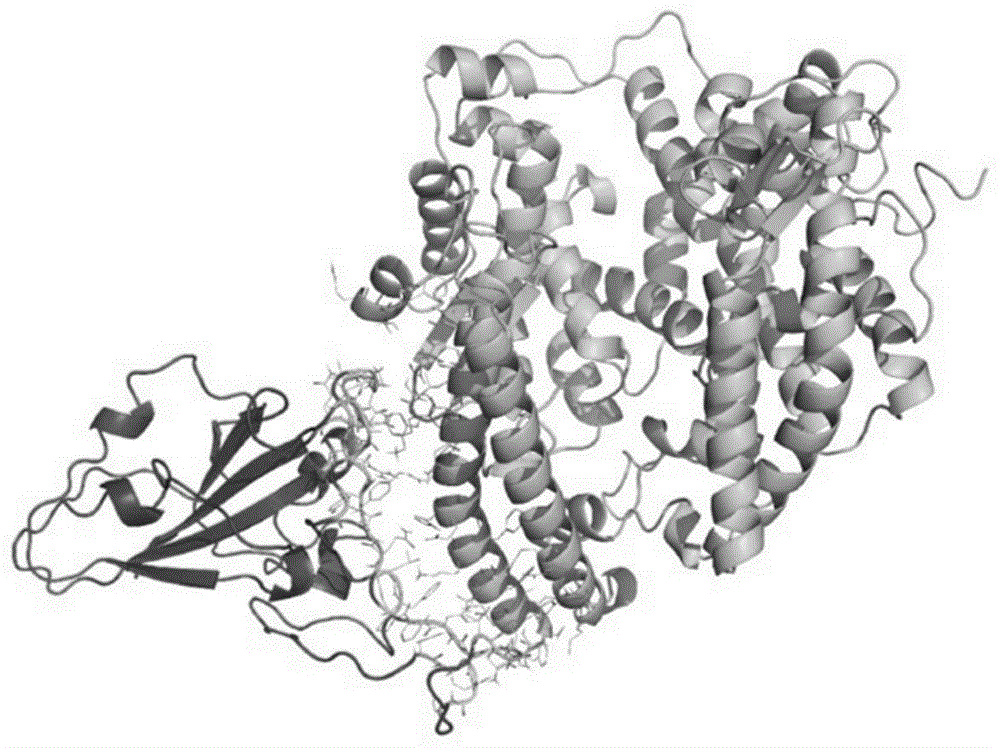
:血管紧张素转化酶2(ace酶2)是ace酶的一种同源蛋白。冠状病毒如新型冠状病毒(sars-cov-2)与非典型性肺炎(sars-cov)和中东呼吸综合征(mers-cov),以ace2酶为受体进入细胞,因此,如果能够阻止ace2酶的表达,有可能降低冠状病毒的感染,由于ace2酶本身具有重要的生理功能,阻止ace2酶的表达,有可能产生无法预期的毒副作用。冠状病毒通过外层包被蛋白-突刺蛋白(spike-蛋白,又称s-蛋白,冠状病毒依靠其刺突蛋白与宿主细胞表面受体结合而入侵细胞)与宿主细胞ace2蛋白结合,如果能够阻止该结合,可能对冠状病毒有很好的抑制效果,得到理想的抗冠状病毒药物。我们采用计算机模拟技术,对已知药物进行了扫描,发现二脒那秦(diminazene)对s-蛋白和ace2蛋白的相互作用界面的有较强的结合能力(对接打分8.0891,见表1),对其结构修饰,我们获得了对s-蛋白和ace2蛋白的相互作用界面的结合能力更强的化合物lj-01(对接打分8.3138,见表1),提示lj-01能够更好的干扰s-蛋白和ace2蛋白的相互作用。我们将lj-01和可能对冠状病毒有效的药物进行链接,希望获得的具有对s-蛋白和ace2蛋白的相互作用界面有更好结合能力的新抗冠状病毒感染的化合物,提高其靶向性,从而提高药效和安全性。选择的可能对冠状病毒有效的药物有:利巴韦林(ribavirin)、galidesivir、瑞德西韦(remdesivir)、gs-441524(瑞德西韦是gs-441524的磷酸酯前药)、阿比多尔(arbidol)、洛匹那韦(lopinavir)、达芦那韦(darunavir)、氯喹(chloroquine)等。我们将lj-01和以上的抗病毒药物以酯键的方式链接,获得的化合物采用计算机模拟其对s-蛋白和ace2蛋白的相互作用界面的结合能力,部分化合物的结合能力(对接打分)表现出显著的提高(见表1)。其中lj-01和利巴韦林、瑞德西韦以酯基的方式链接,获得的化合物lj-02、lj-04结合能力(见表1:对接打分:10.0797、10.5261)表现出显著的提高,提示该2个化合物对s-蛋白和ace2蛋白的相互作用界面有更强的结合能力。考虑到瑞德西韦是gs-441524的磷酸酯前药,我们将lj-01和gs-441524以酯基的方式链接,获得的化合物的结合能力没有表现出显著的提高(见表1:lj-10对接打分:8.1196),将lj-01和利巴韦林的磷酸酯前药以酯基的方式链接,获得的化合物lj-09,结合能力和lj-02相比,同样没有表现出显著的提高(见表1:lj-09对接打分:9.0412)。说明这个链接具有特异性。初步的体内药物代谢研究显示,获得的目标化合物在ace2蛋白高表达的肺部具有更好的分布。考虑到目标化合物还能够更好的干扰s-蛋白和ace2蛋白的相互作用,从而减少冠状病毒的感染,提示该类药物具有更好的药效和安全性。技术实现要素:解决的技术问题:一类n-苄基取代的二脒那秦衍生物及其药物用途,该类药物能够作用于spike与ace2蛋白相互作用界面,增加药物在ace2蛋白高表达的肺部的分布,可用于制备治疗依赖于ace2酶的病毒性感染的药物。技术方案:一类n-苄基取代的二脒那秦衍生物,结构符合通式(i)其中:r为利巴韦林和瑞德西韦。优选结构如下所示:上述一类n-苄基取代的二脒那秦衍生物在制备治疗依赖于ace2酶的病毒性感染的药物中的应用。治疗依赖于ace2酶的病毒性感染的药物,有效成分为上述一类n-苄基取代的二脒那秦衍生物或其药学上可接受的盐。有益效果:该类药物能够作用于spike与ace2蛋白相互作用界面,增加药物在ace2蛋白高表达的肺部的分布,可用于制备治疗依赖于ace2酶的病毒性感染的药物。附图说明图1为通过同源建模得到的2019-ncovs蛋白与受体ace2蛋白的结合模式图。其中深色的是2019-ncovs蛋白(左),浅色的是ace2(右),连接部是两个蛋白中有相互作用的残基(中间空腔部分)。具体实施方式下面的实施例可使本专业技术人员可全面的理解本发明,但不以任何方式限制本发明。实施例1计算机模拟筛选化合物对s-蛋白和ace2蛋白的相互作用界面的结合能力:采用通过同源建模得到的2019-ncovs蛋白与受体ace2蛋白的结合模式。将化合物和该结合模式两个蛋白中有相互作用的残基进行对接,进行打分。打分结果见表1实施例2目标化合物的合成:2.1关键中间体m-01的合成合成路线:操作:100ml茄形瓶中加入对氨基苯腈11.8g(0.1mol),无水乙醇200ml,3-甲醛基苯甲酸15.0g(0.1mol),50℃反应4h。过滤,固体用无水乙醇200ml分散,0℃分批加入硼氢化钠5.0g,加毕,50℃反应1h。用盐酸调ph为3-5,过滤,滤液加入水20ml,过滤收集固体。向上固体加入100ml水,于-5-0℃加入浓硫酸20g,亚硝酸钠20g,于-5-0℃反应1小时。加入亚硫酸钠消除剩余的亚硝酸钠,滴加对氨基苯腈11.8g(0.1mol),继续反应2小时。过滤,得固体。将该固体加入无水乙醇20ml,37%盐酸10ml,室温反应10小时,加入氨水20ml,于-5-0℃反应24小时,室温反应48小时,于0℃通入氯化氢至无固体产生,过滤,得关键中间体m-01。m-01:1hnmr(400mhz,dmso-d6)δ(ppm)8.48(s,1h),8.37(s,1h),7.92-7.66(m,10h),4.24(s,2h)2.2目标化合物1的合成:合成路线:操作:100ml茄形瓶中加入2.44g(10mmol)利巴韦林,无水ch2cl2100ml,冰浴,加入5.63g(1.0mmol)m-01。0℃,滴加dcc2.4g(11.1mmol)溶于50ml无水ch2cl2的溶液,冰浴反应1h后加入1.0gdmap,室温反应12h。滤除析出的dcu,蒸除溶剂,加乙酸乙酯100ml溶解,分别用5%nahco3洗,5%柠檬酸洗,饱和nacl水洗,无水mgso4干燥。蒸除溶剂。柱层析(石油醚-etac=4∶1),得目标化合物1。目标化合物1:1hnmr(400mhz,dmso-d6)δ(ppm)8.87(s,1h),8.45(s,1h),8.32(s,1h),7.90-7.62(m,12h),5.81(d,1h),5.58(d,1h),5.19(d,1h),4.92(t,1h),4.37-4.33(m,1h),4.16-4.11(m,1h),4.25(s,2h),3.97-3.93(m,1h),3.66-3.15(m,1h),3.50-3.47(m,1h)2.3目标化合物2的合成:参考目标化合物1的方法,以瑞德西韦和关键中间体m-01合成。目标化合物2:1hnmr(400mhz,dmso-d6)δ(ppm):8.46(s,1h),8.36(s,1h),7.92-7.65(m,11h),7.34-7.26(m,2h),7.21-7.11(m,3h),6.92(d,1h),6.88(d,1h),4.79(d,1h),4.43-4.33(m,2h),4.30-4.27(m,1h),4.24(s,2h),4.17(t,1h),4.03-4.01(m,1h),3.98-3.84(m,2h),1.49–1.40(m,1h),1.35–1.27(m,8h),0.85(t,6h),0.72(d,3h),.31pnmr(162mhz,dmso-d6)δ(ppm)3.66(s)实施例3:目标化合物在动物体内分布的考察spf级sd大鼠,6-8周龄,体重180-220g,雄性,静脉注射目标化合物1(12.8mg/kg,2.0mmol/l),给药后1小时,测定其血药浓度和肺组织中的浓度,并以利巴韦林为参比(4.88mg/kg,2.0mmol/l)。表2目标化合物1静脉注射给药后1小时后血液、肝、肺中的利巴韦林浓度比较(以利巴韦林为对照)化合物编号血液肝肺10.60.72.8利巴韦林1.01.01.0以上实验结果显示:注射给药目标化合物1后,和直接注射利巴韦林相比,血液、肝中药物的浓度度显著减少,肺组织中药物浓度显著增大。说明目标化合物1对ace2酶高表达的肺具有靶向作用。同法考察了目标化合物2的动物体内分布,结果见表3表3目标化合物2静脉注射给药后1小时后血液、肝、肺中的瑞德西韦(含其代谢物)浓度比较(以瑞德西韦为对照)化合物编号血液肝肺10.50.63.2瑞德西韦及其代谢物1.01.01.0以上实验结果显示:注射给药目标化合物2后,和直接注射瑞德西韦相比,血液、肝中药物的浓度显著减少,肺组织中要物的浓度显著增大。目标化合物1对ace2酶高表达的肺具有靶向作用。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1