微卫星不稳定的预测方法及其应用与流程
本发明属于基因检测
技术领域:
,具体涉及一种微卫星不稳定的检测预测方法,以及相关的位点筛选方法、模型构建方法、标志物组合、试剂盒、系统、装置、计算机可读存储介质、设备,以及相关的微卫星不稳定量化方法。
背景技术:
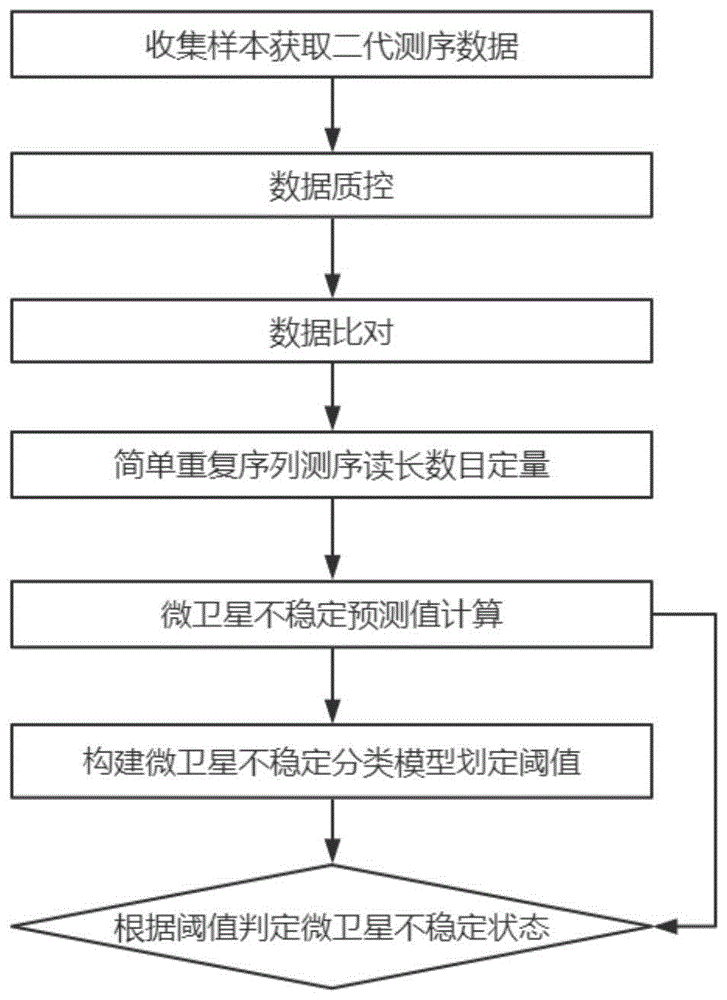
:微卫星(microsatellites)序列是一些简单串联重复区域存在于人类基因组上数百万个基因座(loci)中。微卫星不稳定(microsatelliteinstability,msi)是指由于dna错配修复缺陷(dmmr)导致的微卫星序列变长或变短的现象。这类体细胞突变可导致抑癌基因失活或破坏其他非编码调控序列,从而起到致癌作用。msi发展至今成为临床关注的热点,主要原因在于免疫治疗对临床的冲击。在过去的几年中,癌症疗法已经得到了广泛的发展,人们认识到具有独特分子表型的不同癌症亚型可以使用新型靶向精准治疗。据报道,msi作为这些独特的分子表型之一存在于多种癌症中,包括结直肠癌,子宫内膜癌,胃癌,前列腺癌,卵巢癌和成胶质细胞瘤等。在各类癌症类型中,尤其是结直肠癌中,msi状态,特别是高水平的msi状态(msi-h)已经被认为是预后生物标志物,随后也有研究报道msi状态被做为泛癌种免疫筛查点封锁反应的预后生物标志物,在2017年美国食品药品监督管理局(fda)批准了派姆单抗作为细胞编程性死亡1(pd-1)抑制剂用于治疗具有高水平微卫星不稳定(msi-h)或dmmr而没有其他治疗选择的所有晚期实体瘤患者。临床试验表明,免疫检查点抑制剂可改善多种实体瘤的疗效,这使msi-h和dmmr成为治疗反应的首个全癌预测指标。因此,探究检测msi状态的工具和方法在肿瘤临床诊断和预后治疗上具有重要意义。截至目前msi检测方法主要有三种,免疫组化(immunohistochemicalstains,ihc)、聚合酶链反应(polymerasechainreaction,pcr)和二代测序(next-generationsequencing,ngs)。ihc方法是利用ffpe肿瘤组织切片通过判断4种dna错配修复蛋白(mlh1,pms2,msh2,msh6)的表达情况来明确细胞是否存在错配修复功能缺陷。若结果显示任一蛋白表达缺失,则判读为dmmr;若四个蛋白全部表达,则判读为错配修复功能正常(pmmr)。其优势在于应用性广泛,并能确定哪些mmr蛋白在肿瘤中丢失,然而ihc存在检不到某些定性蛋白的变化,mmr结果偶有报错,以及结果判读因人而异等问题。pcr方法是通过比较肿瘤与非肿瘤组织dna的微卫星序列长度的变化来鉴定肿瘤细胞是否存在dmmr,这种检测方法被广泛认为是msi检测的金标准的诊断工具。通常检测至少5个以上的位点,1个位点不稳定称为低度微卫星不稳定(msi-l),2个及2个以上位点不稳定称为msi-h,5个位点均稳定即微卫星稳定(mss)。msi-l和mss等同于pmmr的概念,msi-h等同于dmmr的概念。pcr检测msi方法不仅弥补了ihc在因非截断式错义突变导致的msi无法检出的漏洞同时还具备良好的可重复性,然而它也有局限性,包括基因组合(panel)的位点较少,通量较低,二核苷酸重复序列引起的msi检测敏感性较低等。ngs方法是基于二代测序对待测样本在全基因组或全外显子组范围上进行msi检测的方法。msi作为生物标记主要作用是用于区分msi突变表型并非是找出确定的msi突变位置,因此,ngs方法弥补了pcr的通量低,panel小的问题,可与靶点突变检测、肿瘤突变负荷(tmb)等检测共用测序数据,检测位点可以根据检测目的自行调整省时省力、灵活方便。在目前已报道的ngs方法中,一般以pcr检测结果作为金标准,比较二者结果一致性作为评价ngs检测方法性能的标准。然而目前ngs方法繁多且多为配对方法,例如已发表的msi检测工具msings、msisensor、mantis等。随着msi在临床上的广泛应用,与pcr和ihg方法相比ngs以其速度快、通量高、以及覆盖检测位点广的优势成为msi临床检测的发展趋势。这就对ngs方法在准确性和可行性有了更高的需求,尤其是panel检测中表现优异的性能要求更加迫切,然而目前报道的ngs方法大多是基于肿瘤组织和对照组织比较的方法检测msi,其检测标准参差不齐,在panel应用中性能表现也并不十分理想,这就限制了ngs方法在临床上的应用。通过思考和研究已有ngs方法的优势并结合panel分析具体案例数据,利用新的算法可以得到一种适用于panel分析基于单肿瘤样本的msi检测方法。技术实现要素:本发明的目的是针对现有技术存在的缺陷,提供一种微卫星不稳定(msi)的检测预测方法,以及相关的位点筛选方法、模型构建方法。发明人发现一种合理的算法拟合msi分布可以极大改善现有ngs(下一代测序)方法检测msi的性能,更重要的是,与现有方法相比,本发明的方法突破了已有ngs方法必须要有对照样本的限制,并且在panel分析应用中性能表现优异,具有更高的灵敏度、特异性和准确性。根据本发明方法的原理,其可嵌入各种已知的或将要开发的基因组合(panel)中,对panel检测范围内的微卫星区域进行简单重复序列定量(repeatcount),基于微卫星区域定量结果利用本发明中的算法拟合每个msi位点的msi预测值并利用随机森林算法对msi检测位点降噪、构建msi分类模型(图1),即可实现准确的预测。为了实现以上目的,本发明提供了一种微卫星不稳定(msi)检测位点或微卫星不稳定检测位点组合的筛选方法,所述筛选方法:i)包含测定样本中候选微卫星位点的微卫星不稳定预测值的步骤;和/或ii)使用包括候选微卫星位点的微卫星不稳定预测值在内的一种或多种指标筛选用于微卫星不稳定检测的位点或位点组合;所述微卫星不稳定预测值是候选微卫星位点中主等位基因(allele)型与次等位基因型之间丰度差异的量化值。在本发明的具体实施方案中,等位基因型的丰度可以通过简单重复序列测序读长数目(repeatcount)计算。在本发明的具体实施方案中,等位基因型的丰度可以通过该基因型读段(reads)支持数计算。任选地,等位基因型的丰度通过归一化落在0-1区间内;优选地,等位基因型丰度归一化的方法可以是,用所述候选微卫星位点中所述等位基因型的读段支持数除以该位点所有等位基因型的读段支持数之和。本发明中,选择重复(repeat)长度在0-50bp之间的等位基因型进行计算。在本发明的具体实施方案中,所述筛选方法还包含通过样本中所有候选微卫星位点的微卫星不稳定预测值计算样本的微卫星不稳定预测值的步骤。进一步地,所述样本的微卫星不稳定预测值按照以下公式计算:其中,f(x)表示样本的微卫星不稳定预测值,xi1表示归一化的主等位基因型丰度,xi2表示归一化的次等位基因型丰度,i为位点编号或位点顺序,n为位点数目。在本发明的具体实施方案中,所述候选微卫星位点可以是全基因组中的微卫星位点、外显子组中的微卫星位点、基因组中一个或多个区域中包含的微卫星位点或者感兴趣的微卫星位点。在本发明的具体实施方案中,所述候选微卫星位点可以选自单核苷酸重复序列、二核苷酸重复序列和/或复杂重复序列。在本发明的具体实施方案中,所述筛选方法还包含通过机器学习对检测位点降噪的方法。进一步地,所述降噪方法可以选自随机森林(randomforest)、主成分分析(pca)、线性判别分析(lda)、岭回归、lasso回归、神经网络或者衍生自上述方法的算法;优选随机森林或者衍生自随机森林的算法。进一步地,所述衍生自随机森林的算法可以选自孤立森林(isolationforest)算法、trte(totallyrandomtreesembedding)算法或者极限树(extratrees)算法。在本发明的具体实施方案中,将微卫星不稳定预测值作为机器学习的输入数据。本发明的筛选方法可以仅使用来源于肿瘤患者或肿瘤组织的样本;本发明的筛选方法可以不使用来源于健康个体或正常组织的样本。本发明还提供一种微卫星不稳定(msi)检测模型的构建方法,所述构建方法:i)包含测定样本中候选微卫星位点的微卫星不稳定预测值的步骤;和/或ii)使用包括候选微卫星位点的微卫星不稳定预测值在内的一种或多种指标构建微卫星不稳定检测模型;所述微卫星不稳定预测值是候选微卫星位点中主等位基因型与次等位基因型之间丰度差异的量化值。在本发明的具体实施方案中,等位基因型的丰度通过简单重复序列测序读长数目(repeatcount)计算。在本发明的具体实施方案中,等位基因型的丰度通过该基因型读段(reads)支持数计算。任选地,等位基因型的丰度通过归一化落在0-1区间内;优选地,等位基因型丰度归一化的方法是,用所述候选微卫星位点中所述等位基因型的读段支持数除以该位点所有等位基因型的读段支持数之和。在本发明的具体实施方案中,可以选择重复(repeat)长度在0-50bp之间的等位基因型进行计算。在本发明的具体实施方案中,所述构建方法还包含通过样本中所有候选微卫星位点的微卫星不稳定预测值计算样本的微卫星不稳定预测值的步骤。进一步地,所述样本的微卫星不稳定预测值可以按照以下公式计算:其中,f(x)表示样本的微卫星不稳定预测值,xi1表示归一化的主等位基因型丰度,xi2表示归一化的次等位基因型丰度,i为位点编号或位点顺序,n为位点数目。在本发明的具体实施方案中,所述候选微卫星位点可以是全基因组中的微卫星位点、外显子组中的微卫星位点、基因组中一个或多个区域中包含的微卫星位点或者感兴趣的微卫星位点。在本发明的具体实施方案中,所述候选微卫星位点可以选自单核苷酸重复序列、二核苷酸重复序列和/或复杂重复序列。在本发明的具体实施方案中,所述构建方法还可以包含通过机器学习对模型所使用的检测位点降噪的方法。进一步地,所述降噪方法可以选自随机森林(randomforest)、主成分分析(pca)、线性判别分析(lda)、岭回归、lasso回归、神经网络或者衍生自上述方法的算法;优选随机森林或者衍生自随机森林的算法。进一步地,所述衍生自随机森林的算法可以选自孤立森林(isolationforest)算法、trte(totallyrandomtreesembedding)算法或者极限树(extratrees)算法。在本发明的具体实施方案中,将微卫星不稳定预测值作为机器学习的输入数据。本发明的构建方法可以仅使用来源于肿瘤患者或肿瘤组织的样本;本发明的构建方法可以不使用来源于健康个体或正常组织的样本。在本发明中,还提供一种微卫星不稳定(msi)检测模型的构建方法,所述构建方法使用本发明的筛选方法筛选得到的微卫星不稳定检测位点或检测位点组合构建模型。进一步地,所述构建方法可以是机器学习方法。本发明还提供一种微卫星不稳定的检测或预测方法,所述方法:i)包含本发明的筛选方法筛选微卫星不稳定检测位点或检测位点组合的步骤;和/或ii)使用本发明的筛选方法筛选得到的微卫星不稳定检测位点或检测位点组合进行检测;和/或iii)使用本发明的构建方法构建得到的微卫星不稳定检测模型进行检测。本发明还提供微卫星不稳定检测标志物组合,所述标志物组合是本发明的筛选方法获得的检测位点处的微卫星标志物组合。进一步地,本发明的检测标志物组合可以包含表2、表5、表6或表7中的标志物。本发明还提供用于特异性检测微卫星不稳定检测位点或检测位点组合的试剂在制备微卫星不稳定检测试剂盒和/或肿瘤伴随诊断试剂盒中的用途,所述微卫星不稳定检测位点或检测位点组合是采用本发明的筛选方法得到的微卫星不稳定检测位点或检测位点组合,或者是本发明所述的检测标志物组合,或者是本发明的构建方法构建的检测模型中的微卫星不稳定检测位点或检测位点组合。本发明还提供一种微卫星不稳定检测试剂盒和/或肿瘤伴随诊断试剂盒,所述试剂盒包含用于特异性检测微卫星不稳定检测位点或检测位点组合的试剂,所述微卫星不稳定检测位点或检测位点组合是采用本发明的筛选方法得到的微卫星不稳定检测位点或检测位点组合,或者是本发明所述的检测标志物组合,或者是本发明的构建方法构建的检测模型中的微卫星不稳定检测位点或检测位点组合。本发明还提供一种用于微卫星不稳定检测和/或肿瘤伴随诊断的系统或装置,所述系统或装置包括:获取模块,用于获取受试者的微卫星不稳定检测位点或检测位点组合的测定数据,所述微卫星不稳定检测位点或检测位点组合是采用本发明的筛选方法得到的微卫星不稳定检测位点或检测位点组合,或者是本发明所述的检测标志物组合,或者是本发明的构建方法构建的检测模型中的微卫星不稳定检测位点或检测位点组合,所述测定数据是检测位点的微卫星不稳定预测值,所述微卫星不稳定预测值是候选微卫星位点中主等位基因型与次等位基因型之间丰度差异的量化值;数据分析模块,用于将所述微卫星不稳定检测位点或检测位点组合的测定数据输入本发明的构建方法构建的检测模型中,以得出检测结果。在本发明的具体实施方案中,所述系统或装置还可以包括:测序模块,用于对受试者进行测序。在本发明的具体实施方案中,所述系统或装置还包括:诊断模块,用于生成肿瘤伴随诊断结果和/或处置建议。本发明还提供一种计算机可读存储介质,所述计算机可读存储介质包括存储的计算机程序,所述计算机程序包含:i)用于执行本发明的微卫星不稳定检测位点或微卫星不稳定检测位点组合的筛选方法的程序;和/或ii)用于执行本发明的微卫星不稳定检测模型的构建方法的程序;和/或iii)用于执行本发明的微卫星不稳定的检测或预测方法的程序。本发明还提供一种设备,包括处理器、存储器以及存储在所述存储器中的计算机程序,所述计算机程序包括:i)用于执行本发明的微卫星不稳定检测位点或微卫星不稳定检测位点组合的筛选方法的程序;和/或ii)用于执行本发明的微卫星不稳定检测模型的构建方法的程序;和/或iii)用于执行本发明的微卫星不稳定的检测或预测方法的程序。本发明还提供一种微卫星不稳定的量化方法,其使用微卫星不稳定预测值对所述微卫星不稳定进行量化,所述微卫星不稳定预测值是微卫星位点中主等位基因型与次等位基因型之间丰度差异的量化值。在本发明的具体实施方案中,等位基因型的丰度通过简单重复序列测序读长数目(repeatcount)计算。在本发明的具体实施方案中,等位基因型的丰度通过该基因型读段(reads)支持数计算。任选地,等位基因型的丰度通过归一化落在0-1区间内;优选地,等位基因型丰度归一化的方法是,用所述微卫星位点中所述等位基因型的读段支持数除以该位点所有等位基因型的读段支持数之和。在本发明的具体实施方案中,可以选择重复(repeat)长度在0-50bp之间的等位基因型进行计算。在本发明的具体实施方案中,所述量化方法还包含通过样本中所有目标微卫星位点的微卫星不稳定预测值计算样本的微卫星不稳定预测值的步骤。进一步地,所述样本的微卫星不稳定预测值按照以下公式计算:其中,f(x)表示样本的微卫星不稳定预测值,xi1表示归一化的主等位基因型丰度,xi2表示归一化的次等位基因型丰度,i为位点编号或位点顺序,n为位点数目。在本发明的具体实施方案中,所述微卫星位点是全基因组中的微卫星位点、外显子组中的微卫星位点、基因组中一个或多个区域中包含的微卫星位点或者感兴趣的微卫星位点。在本发明的具体实施方案中,所述微卫星位点选自单核苷酸重复序列、二核苷酸重复序列和/或复杂重复序列。本发明的各项方法还可以包含测序的步骤,测定样本的核苷酸序列。本发明的各项方法可以仅使用来源于肿瘤患者或肿瘤组织的样本;本发明的方法可以不使用来源于健康个体或正常组织的样本。本发明的技术方案可以在癌症的各种诊断和非诊断的应用场景中使用。本发明的技术方案可适用于任何分期的肿瘤,例如极早期肿瘤、早期肿瘤、中期肿瘤、晚期肿瘤;优选用于早期肿瘤或极早期肿瘤。本发明的有益效果至少包括以下方面:(1)准确性高,模型性能优良。灵敏度、特异性和auc可以达到1,预测结果与作为金标准的pcr方法完全一致,在检测限测lod20%、lod15%、lod10%时检出率均能达到100%,相比与现存ngs方法性能更加优良。(2)仅基于单肿瘤样本就可以精准稳定地检测msi状态,摆脱了因对照样本遗失或取样困难所带来的限制,同时在技术层面上也节省了测序成本、简化了操作步骤、提高了检测速度。(3)本发明方法能灵活的嵌入不同基因检测panel产品的检测流程,可以根据panel大小灵活筛选msi检测位点,并具有优异的msi检测性能,应用灵活广泛。对于全新的panel产品,可以根据panel检测范围选取其中所有的msi检测位点,采用本发明的方法筛选出其中有效的检测位点,使用本发明的算法和模型对该panel的样本做msi预测。对于其他panel产品,如果其检测范围包含于已经采用本发明方法筛选过的已有panel中,可以直接从已有panel筛选出的检测位点中提取该panel范围内的位点,即可用于msi预测;对于检测范围与已有panel有差集的panel产品,可采用本发明的方法从头筛选出高效位点、构建预测模型。附图说明图1为本发明实施例中的微卫星不稳定预测流程技术路线图。图2为本发明实施例中的微卫星不稳定分类模型的构建及性能评估流程图。图3为本发明模型的性能评估结果(训练集样本)。左图为模型roc曲线;右图为分类散点图。图4为本发明模型的性能评估结果(158例样本)。左图为数据汇总;右图为分类散点图。具体实施方式如无特别指明,本发明所使用术语均具有本领域通常的含义,所使用的试剂均为本领域常规商业化试剂。本发明中术语“基因型读段(reads,或称读长)支持数”是指测序获得的读段与某一种基因型的序列相一致的数目。对于微卫星不稳定而言,其基因型是染色体同一位置存在的简单串联重复序列种类,每一个简单串联重复序列种类称作一个基因型。术语“简单重复序列测序读长数目”(repeatcount)就是指每种简单重复序列的读段支持数。本发明中术语“灵敏度”(sensitivity)可以指真阳性的数量除以真阳性与假阴性数量的总和,可以用来表征正确鉴别真正患有癌症的群体的能力。本发明中术语“特异性”(specificity)可以指真阴性的数量除以真阴性与假阳性数量的总和,可以用来表征正确鉴别真正未患癌症的群体的能力。本发明中术语“roc”或“roc曲线”可以指受试者工作特征曲线(receiveroperatingcharacteristiccurve),可以用来表征分类器的表现。可以通过在各个阈值设置下用灵敏度对特异性作图来生成roc曲线。本发明中术语“auc”可以指roc曲线下的面积,可以用来表征癌症筛查/预测的表现。auc的范围为0.5-1.0,数值越接近1.0表明该方法的筛查/预测表现越好。下面将结合附图和具体实施例对本发明的技术内容作详细说明。本领域技术人员将会理解,以下实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例1收集临床患者组织样本,包括肿瘤待测样本及其配对的健康人对照(normal)样本,用于测试本发明的msi检测方法,并用pcrmsi检测方法对其进行验证,并与其他现有技术已知的msi检测工具进行比较。其中,以pcrmsi检测结果作为金标准,比较其与本发明的msi检测方法的结果一致性,用于评价本发明的msi检测方法的性能。需要说明的是,本发明提出的msi检测方法并不需要使用对照样本,对照样本在实施例部分用于pcr验证和其他msi检测软件分析。至于用于测试的msi位点,本实施例中使用了一个大型基因组合(panel)进行测试,该panel包含654个与直肠癌、胃癌、卵巢癌和肺肉瘤癌等肿瘤相关的基因,覆盖了550个msi位点。pcr方法检测msi所用试剂盒为promega试剂盒,pcr检测得到每例样本的msi状态信息,将其结果作为金标准。将pcr的结果分为两组:检测为msi-h(微卫星高度不稳定)的样本为一组,记作msi-h(表示阳性);检测为msi-l(微卫星低度不稳定)和mss(微卫星稳定)的样本为一组,记作mss(表示阴性)。这样分组的目的是将pcr检测结果与基于ngs的msi检测方法的结果进行对应(ngs检测方法在现有技术报道中的检测结果只有msi-h和mss两种)。实施例2对实施例1中的4个基因组合进行外显子探针捕获建库测序,使用基因测序仪(nextseqcn500),按照仪器标准操作规程进行150bppair-end模式测序(read1:151,read2:151;index1:8,index2:8),最终得到fastq格式二代测序数据作为原始数据(rawdata)。利用质控软件fastp对得到的二代测序数据进行质量控制,过滤测序接头、低质量碱基以及测序错误片段等,过滤后获得高质量的数据(cleandata)。使用比对软件bwa用cleandata与参考基因组hg19比对,得到每个dna片段基因组上对应的具体位置信息;再使用gencore软件对比对数据进行去重复、碱基校正处理。根据已知参考基因组检测区域的重复(repeat)位置,获取每种repeat类型的读段(reads,或称读长)支持数。首先,利用已发表软件mantis自带工具repeatfinder生成基因组msi候选位点bed文件,从bed文件中读取位点(loci),利用提供的参考基因组文件来对比校正0-/1-碱基索引产生的差异。其中,选取了长度在10-100之间、单碱基、重复次数在5次以上的微卫星重复序列。其次,对每个loci上所有repeat类型匹配到的reads进行质控,每条reads的平均碱基质量不小于20,每条reads落在loci区间内的平均碱基量不小于25,reads长度(只统计非剪切部分,不统计soft-clipped或hard-clipped部分)大于35bp,最小repeatreads支持数大于1,涉及到的其他过滤步骤均为软件默认参数。最后,分别统计肿瘤患者和对照的所有位点每种repeat类型对应质控合格的reads数,生成简单重复序列测序读长数目(repeatcount)结果用于后续分析(由于mantis是分析配对样本的,需要同时输入肿瘤和对照样本,而本发明的方法并不需要使用对照样本,因而为了不影响软件运行,将肿瘤样本当作对照样本重复计数一次,最终使用的是肿瘤样本的统计结果)。实施例3根据实施例2中获得的repeatcount结果,在每个位点上获得所有repeat类型代表该位置的等位基因型,对于每一个loci计算主allele和次allele丰度值之差作为该loci微卫星不稳定水平预测值,样本中所有loci的(1-不稳定水平预测值)的平均值作为样本的微卫星不稳定预测值(msiscore),无参算法计算公式如下:其中,xi1表示主峰值,xi2表示次峰值,i为loci顺序,n为loci数目。具体地,统计每个检测位置repeat长度在0-50bp之间的repeatcount,对repeatcount进行归一化使之落在0-1区间内,归一化的方式为该位点每一个repeat类型的reads支持数除以该位点所有repeat类型的reads支持数之和,根据归一化后的数值按照从大到小排序,最大值为主峰值,次值为次峰值。实施例4将实施例1中收集的样本中的153例肿瘤组织样本按照8:2的比例和pcr方法测得的msi状态信息(msi-h/mss)随机分层抽样划分为训练集122例和测试集31例,接着将初始训练集样本进一步按照训练集:验证集为8:2的比例和msi状态信息(msi-h/mss)随机分层抽样20次,得到20次抽样结果的样本(图2),每次抽样得到训练集样本97例、验证集样本25例。上述样本中不含任何对照样本。对每次抽样样本进行以下操作,针对每例样本以实施例1中大型panel所覆盖的550个候选位点,使用实施例3中的无参算法计算msiscore,再将每次抽样样本集合整理成训练集和验证集的矩阵,该矩阵包含每个样本在每个检测位点的msiscore。对于每次抽样集合整理的矩阵,利用随机森林算法计算出每个候选位点的权重,将所有候选位点按照权重在20次抽样中不为0的次数排序,按照权重出现次数大于等于0-9划分10个梯度(如表1所示)。表1权重梯度划分结果权重出现次数>=0>=1>=2>=3>=4>=5>=6>=7>=8>=9检测位点数550381265200156129108988371根据每个梯度选取的检测位点构建模型对训练集进行预测分类,用训练集划分阈值并对验证集进行预测分类,用验证集的预测分类效果评估本次评估模型性能。根据每次梯度测试的结果,绘制roc曲线评估每个模型性能,兼顾分类性能和稳定性选择权重出现次数>=3的梯度所包含的200个检测位点的模型,根据训练集在选定候选位点的测试结果划定阈值。检测位点选取结果如下表所示:表2检测位点筛选结果表2(续)表2(续)针对97例训练集样本预测数据绘制roc曲线评估模型性能,结果如图3所示,采用本发明方法构建的模型,其灵敏度、特异性和auc均为1,实现了对msi的准确预测。进一步地,还使用了158例样本评估模型性能表现,msiscore>=阈值判定为msi-h,反之为mss。结果如表3和图4所示:表3模型预测性能:158例样本中的msi-h样本均被本发明构建的模型正确检出,其与pcr检测结果完全一致,二者一致性高达100%。为了验证本发明方法最高灵敏度范围,我们收集了msi阳性(msi-h)参考品km12和msi阴性(mss)参考品na12878,按比例混合msi-h细胞系和mss细胞系,稀释msi-h细胞系浓度(分别稀释为20%、15%、10%、5%),用以确定最低msi检测限。使用实施例1中的大型panel作为测序panel,对上述细胞样本上机测序检测msi。结果如表4所示,在lod20%、lod15%、lod10%的情况下,msi-h检出率均能达到100%;在lod5%的情况下,msi-h检出率仍能达到83%。表4不同lod下的检出率:稀释浓度(%)2015105检出率(%)10010010083实施例5为了评估模型的泛化性能,引入了中型、小型、微型三个基因panel,分别包含与直肠癌、胃癌、卵巢癌和肺肉瘤癌等肿瘤相关的基因457个、86个、31个,分别覆盖msi位点146个、109个、41个。分别使用上述三个panel对173例新的样本进行上机测序。实施例4中筛选得到的200个msi位点中,分别有61、33、7个位点在中型、小型、微型panel中出现,结果如表5-6以及表7中的序号1-7所示。表5检测位点筛选结果(中型panel):表5(续)表6检测位点筛选结果(小型panel):表6(续)表7检测位点筛选结果(微型panel):使用上述位点对173例新样本的测序数据进行分析,结果如表8所示。对于中型、小型和微型panel,直接使用已经筛选出的61个、33个或7个位点均能准确检测出173个样本中的全部11个msi-h样本,灵敏度和特异性均达到1。表8中型、小型、微型panel中模型预测性能:实施例6为了测试本发明的方法相比于现有技术方法所产生的性能改善,与现有技术中的msi检测模型进行了比较。现有技术中,mantis、msisensor和msings是最常用的三种msi检测方法,其中,mantis的灵敏度和特异性被认为是最佳的(performanceevaluationforrapiddetectionofpan-cancermicrosatelliteinstabilitywithmantis.oncotarget,2017,vol.8,(no.5),pp:7452-7463),因而选择mantis作为比较例,基于前述173份样本在3个panel中的测序数据对msi进行预测,mantis软件所用msi检测位点为所述3个panel基因区域所覆盖的所有msi检测位点。结果如表9所示:表9中型、小型、微型panel中模型预测性能:由此可见,采用本发明的方法构建msi检测模型,与现有技术方法相比,能够在使用更少的检测位点的情况下,获得更高的灵敏度和特异性。最后需要说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1