一种弹性蛋白酶抑制剂前药及其用途的制作方法
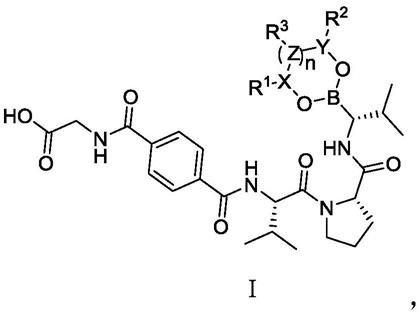
1.本发明属于化合物领域,具体涉及一种取代硼酸类化合物的前药,具体涉及取代硼酸类化合物的前药的混合物或者组合物,尤其是作为弹性蛋白酶抑制剂的硼酸类化合物的前药的混合物或者组合物,用于治疗各种由于弹性蛋白酶过量引发的肺部疾病,包括慢性阻塞性肺病(copd)。
背景技术:
2.人弹性蛋白酶(hne)是一个32kda大小的丝氨酸蛋白酶。很多疾病的病理过程都涉及到该蛋白酶。过量的弹性蛋白酶对人体的破坏性非常大,以至于肝脏会专门合成分泌一种天然抑制剂(α-1抗胰蛋白酶)来保持人体中弹性蛋白酶活性的平衡。弹性蛋白酶在中性白细胞中合成之后被存储在嗜天青颗粒中,直到中性白细胞被外界信号激活后才分泌出来(takahashi h,nukiwa t,basset p,crystal rg.j biol chem.1988;263(5):2543-2547)。它的主要作用是降解弹性蛋白。弹性蛋白是细胞外基质蛋白的主要构成成分之一,其他构成成分包括纤粘蛋白,层粘蛋白,蛋白多糖,iii型和iv型胶原蛋白(bieth,g.in regulation of matrix accumulation,mecham,r.p.(eds),academic press,ny,usa,1986,217-306)。弹性蛋白酶的作用包括通过降解细菌结构蛋白来对抗细菌入侵。总之,人弹性蛋白酶通过降解破损的组织或者入侵的细菌等方式,对于维持机体稳态起到不可或缺的作用。
3.α-1抗胰蛋白酶缺乏症(a1ad)是一种遗传疾病,表征为弹性蛋白酶的天然抑制剂α-1抗胰蛋白酶缺乏后,失去平衡而过量的蛋白酶开始破坏细支气管和肺泡上皮细胞的细胞外基质(laurell and eriksson,1963,scand.j clin.invest.15,132-140)。其病理过程有力地证实了过量弹性蛋白酶活性与慢性肺损伤疾病间的因果关系,尤其是气流受限和肺气肿等病症。在另外一种遗传病囊性纤维化中,失活的cftr蛋白导致了气管中过于黏稠的黏液和慢性炎症反应,进而产生过量积累的弹性蛋白酶,最终也使肺组织结构受损,功能受限。
4.除了上述两种遗传病,弹性蛋白酶也在其他几种慢性肺病中起到关键作用,包括肺纤维化,肺炎,急性呼吸窘迫综合征,慢性细支气管炎,肺气肿等等。其中因为长期气管炎症导致气流受限,还有肺气肿等导致的呼吸困难等病症的慢性肺病统称为慢阻肺(copd)。
5.慢阻肺是一种发病率很高的疾病,目前在中国有大约1亿的慢阻肺患者,而且因为人口老龄化和空气污染等因素,这个数字还在不断增加。该病目前没有有效治疗手段,目前采用的支气管扩张剂(laba,lama)只能缓解症状却不能阻止病程进展。这些病人仍然会不断地发生肺部感染,而且每次炎症之后,肺功能损伤的程度都会加重。
6.综上所述,弹性蛋白酶抑制剂对以上几种疾病都可能产生疗效。目前已经有几种抑制剂进入过临床实验,或者正在临床实验中,以测试对慢阻肺,囊性纤维化或者α-1抗胰蛋白酶缺乏症等疾病的治疗效果。但这些临床实验要么失败,要么仍然未出结果。其中失败的大部分原因并非活性差,而是因为毒性(与弹性蛋白酶共价结合),药代动力学(没有肺部
富集)等原因。专利wo2018175173a1提及了含硼化合物52,其结构为使用创新化学方法合成且与弹性蛋白酶可逆共价结合,是一种新型弹性蛋白酶抑制剂,具有肺部富集率高等优秀特性。然而,动物体内药代动力学研究发现该化合物在肺部暴露时间太短,消除快,可能会限制其药效。因此需要开发一类具有体内暴露时间更长,暴露量更高的弹性蛋白酶抑制剂。
技术实现要素:
7.发明要解决的问题
8.为了解决上述技术问题,本发明提供了相对于专利wo2018175173a1中化合物52具有更高肺部暴露量,更长半衰期的一系列前药化合物。
9.用于解决问题的方案
10.为了解决上述技术问题,本发明提供了以下技术方案:
11.一种具备式ⅰ结构的化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物:
[0012][0013]
其中,
[0014]
当n为0时,
[0015]
x和y直接相连,x和y分别为cr4和cr5,r1和r2独立地选自氢、氘、卤素、c
1-6
烷基和c
3-8
环烷基,或者r1和r2相连形成未取代或被r6取代的c
3-8
环烷基;
[0016]
r4和r5各自独立地选自氢、氘和c
1-6
烷基;
[0017]
每一个r6独立地选自氢、氘、羟基、氨基、卤素、c
1-6
烷基、卤素取代的c
1-6
烷基、c
1-6
烷氧基;
[0018]
或者当n为1-5的整数时,
[0019]
x-r1和y-r2各自独立地选自羰基、chr7,r7选自氢、氘和c
1-6
烷基;
[0020]
每一个z独立地选自o、s、ch和n,每一个r3独立地为不存在、氢、氘和c
1-6
烷基。
[0021]
本发明还提供了一种药物组合物,其包含上述任一化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物。
[0022]
本发明还提供了上述任一化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,或者上述药物组合物,在制备预防和/或治疗弹性蛋白酶介导疾病的药物中的应用,优选在制备预防和/或治疗慢性阻塞性肺炎的药物中的应用。
[0023]
本发明还提供了上述任一所述的化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,或者上述药物组合物,其用作预防和/或治疗弹性蛋白酶介导的疾病的药物,优选用作预防和/或治疗慢性阻塞性肺病的药物。
[0024]
本发明还提供了一种预防和/或治疗弹性蛋白酶介导的疾病的方法,其包括下列步骤:将治疗有效量的根据上述任一化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,或者上述药物组合物施用于对其有需求的患者。
[0025]
发明的效果
[0026]
本发明的化合物相对于专利wo2018175173a1中的化合物52(对照物),具有肺部暴露量高,半衰期长的优点。本发明的前药化合物大大提高了活性化合物(对照物)在肺部的浓度和停留时间,相对于对照物在体内药代动力学方面具有显著改进。
附图说明
[0027]
图1实施例1化合物在37℃模拟肺液中的稳定性数据;
[0028]
图2实施例2化合物在37℃模拟肺液中的稳定性数据;
[0029]
图3实施例1化合物室温条件下在不同溶剂中稳定性;
[0030]
图4实施例2化合物室温条件下在不同溶剂中稳定性;
[0031]
图5对照物小鼠气管内给药(2mg/kg)后肺部药物浓度;
[0032]
图6实施例1化合物小鼠气管内给药(2mg/kg)后在肺部中的实施例1化合物药物浓度和水解产物对照物药物浓度。
具体实施方式
[0033]
为了更为清晰的描述本发明的内容,本技术中所涉及的全部术语定义如下:
[0034]
术语“c
1-6
烷基”单独或者以组合方式表示包含1-6个、特别是1-4个碳原子的饱和直链或支链的烷基,包括甲基、乙基、丙基、异丙基、丁基、仲丁基、异丁基、叔丁基、正戊基、2-戊基、3-戊基、2-甲基-2-丁基、3-甲基-2-丁基、3-甲基-1-丁基、2-甲基-1-丁基、正己基、2-己基、3-己基、2-甲基-2-戊基、3-甲基-2-戊基、4-甲基-2-戊基、3-甲基-3-戊基、2-甲基-3-戊基、2,3-二甲基-2-丁基、3,3,-二甲基-2-丁基等。优选地,“c
1-6
烷基”是甲基、乙基、异丙基、叔丁基中的任一种。
[0035]
术语“c
3-8
环烷基”单独或者以组合方式表示具有3到8个、特别是5-7个碳原子的饱和环烷基,所述c
3-8
环烷基包括单环、二环、桥环和螺环,具体地,所述单环包括环丙基、环丁基、环戊基、环己基、环庚基、环辛基等;所述二环包括二环[2.2.0]己基,二环[3.1.0]己基,二环[3.2.0]庚基,二环[3.3.0]辛基,二环[4.1.0]庚基,二环[4.2.0]辛基等;所述桥环包括二环[2.2.1]庚基,二环[2.2.2]辛基,二环[3.1.1]庚基,二环[3.2.1]辛基,二环[4.1.1]辛基等;所述螺环包括螺[2.2]戊基,螺[2.3]己基,螺[3.3]庚基,螺[2.4]庚基,螺[3.4]辛基,螺[2.5]辛基。特别的,“c
5-8
环烷基”是二环[2.2.2]辛基,二环[3.1.1]庚基等。
[0036]
术语“c
1-6
烷氧基”单独或者以组合方式表示基团c
1-6
烷基-o-,其中“c
1-6
烷基”表示
如以上所定义。
[0037]
术语“氨基”单独或者以组合方式表示伯氨基(-nh2)、仲氨基(-nh-)或叔氨基
[0038]
术语“羟基”单独或者以组合方式表示基团-oh。
[0039]
术语“卤素”单独或者以组合方式表示氟、氯、溴或碘。特别的是氟、氯或溴。
[0040]
术语“异构体”包含所有的同分异构形式包括对映异构体、非对映异构体和几何异构体包括顺反异构体。因此,本发明中所设计的化合物的单个立体化学异构体或其对映异构体、非对映异构体、或几何异构体(或顺反异构体)的混合物都属于本发明的范围。
[0041]
术语“药学上可接受的盐”表示本发明的化合物以它们的药用盐的形式存在,包括酸加成盐和碱加成盐。药学上可接受的盐在s.m.berge在j.pharmaceutical sciences(66卷:1-19页,1977年)中描述的pharmaceutically salts中有所描述。在本发明中,药学上可接受的无毒的酸加成盐表示本发明中的化合物与有机或无机酸形成的盐,有机或无机酸包括但不限于盐酸、硫酸、氢溴酸、氢碘酸、磷酸、硝酸、高氯酸、乙酸、草酸、马来酸、富马酸、酒石酸、苯磺酸、甲磺酸、水杨酸、琥珀酸、柠檬酸、乳酸、丙酸、苯甲酸、对甲苯磺酸、苹果酸等。药学上可接受的无毒的碱加成盐表示本发明中的化合物与有机或无机碱所形成的盐,包括但不限于碱金属盐,例如锂、钠或钾盐;碱土金属盐,例如钙或镁盐;有机碱盐,例如通过与含n基团的有机碱形成的铵盐或n
+
(c
1-6
烷基)4盐。“药学上可接受的盐”可通过一般的化学方法合成。
[0042]
术语“酯”表示本发明化合物通过其结构中的一个或多个羟基与羧酸、磷酸、碳酸、磺酸、硼酸等质子酸中的任意一种或多种形成的酯,或者本发明化合物通过其结构中的一个或多个羧基与醇和/或酚形成的酯。
[0043]
术语“溶剂化物”表示一个或多个溶剂分子与本发明中的化合物所形成的缔合物。形成溶剂化物的溶剂包括但不限于水、甲醇、乙醇、异丙醇、乙酸乙酯、四氢呋喃、n,n-二甲基甲酰胺、二甲亚砜等。
[0044]
术语“水合物”是指水与本发明中的化合物形成的缔合物
[0045]
术语“前药”表示作为本发明的化合物的化学衍生物,该衍生物在体内通过发生化学反应转换成通式i所表示的化合物。
[0046]
术语“同位素衍生物”表示通式i中的氢原子被1-6个氘原子所取代得到的同位素衍生物、通式i中的碳原子被1-3个碳14原子所取代得到的同位素衍生物。
[0047]
以上对本发明的涉及的术语进行了定义,本领域技术人员还可以结合现有技术对以上术语进行理解,以下基于本发明的内容以及对术语的定义进一步进行描述。
[0048]
本发明提供了一种具备式ⅰ结构的化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物:
[0049][0050]
其中,
[0051]
当n为0时,
[0052]
x和y直接相连,x和y分别为cr4和cr5,r1和r2独立地选自氢、氘、卤素、c
1-6
烷基和c
3-8
环烷基,或者r1和r2相连形成未取代或被r6取代的c
3-8
环烷基;
[0053]
r4和r5各自独立地选自氢、氘和c
1-6
烷基;
[0054]
每一个r6独立地选自氢、氘、羟基、氨基、卤素、c
1-6
烷基、卤素取代的c
1-6
烷基、c
1-6
烷氧基;
[0055]
或者当n为1-5的整数时,
[0056]
x-r1和y-r2各自独立地选自羰基、chr7,r7选自氢、氘和c
1-6
烷基;
[0057]
每一个z独立地选自o、s、ch和n,每一个r3独立地为不存在、氢、氘和c
1-6
烷基。
[0058]
在一项优选的实施方案中,当n为0时,x和y直接相连,x和y分别为cr4和cr5,r1和r2相连形成未取代或被r6取代的c
5-8
环烷基,所述c
5-8
环烷基包含单环、二环、桥环和螺环。
[0059]
在一项优选的实施方案中,x和y分别为cr4和cr5,所述r4和r5各自独立地选自氢、氘、甲基和乙基,所述c
5-8
环烷基为或
[0060]
在一项优选的实施方案中,r1和r2相连形成未取代或被1-2个r6取代的c
5-8
环烷基,每一个r6独立地选自氢、氘、羟基、氨基、氟、氯、溴、甲基、乙基、正丙基、异丙基、氟甲基、二氟甲基、三氟甲基、甲氧基和乙氧基。
[0061]
在一项优选的实施方案中,当n为1-5的整数时,x-r1和y-r2均为羰基。
[0062]
在一项优选的实施方案中,当n为1-5的整数时,每一个z独立地选自ch和n,每一个r3独立地选自氢、氘、甲基、乙基、正丙基和异丙基。
[0063]
在一项优选的实施方案中,n为3,每一个z-r3独立地选自亚甲基和氨基甲基,或者n为1,z-r3为亚甲基。
[0064]
在一项优选的实施方案中,所述为其中z1为o、s、n,每一个r3独立地为不存在、氢、氘、甲基或乙基,优选的,z1为n,每一个r3独立地为氢、氘、甲基或
乙基。
[0065]
在一项优选的实施方案中,当n为0时,
[0066]
x和y直接相连,x和y分别为cr4和cr5,r1和r2相连形成未取代或被r6取代的c
5-8
环烷基,所述c
5-8
环烷基为或
[0067]
r4和r5各自独立地选自氢、氘、甲基和乙基;
[0068]
每一个r6独立地选自氢、氘、甲基和乙基;
[0069]
或者当n为3时,
[0070]
x-r1和y-r2均为羰基;
[0071]
每一个z-r3独立地选自亚甲基和氨基甲基。
[0072]
本发明还提供了一种化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,所述化合物为以下任一种:
[0073][0074]
本发明还提供了一种药物组合物,其包含上述任一化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物。
[0075]
本发明还提供了上述任一化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,或者上述药物组合物,在制备预防和/或治疗弹性蛋白酶介导疾病的药物中的应用,优选在制备预防和/或治疗慢性阻塞性肺炎的药物中的应用。
[0076]
本发明还提供了上述任一所述的化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,或者上述药物组合物,其用作预防和/或治疗弹性蛋白酶介导的疾病的药物,优选用作预防和/或治疗慢性阻塞性肺病的药物。
[0077]
本发明还提供了一种预防和/或治疗弹性蛋白酶介导的疾病的方法,其包括下列步骤:将治疗有效量的根据上述任一化合物或其药学上可接受的盐、酯、异构体、溶剂化物、前药或同位素标记物,或者上述药物组合物施用于对其有需求的患者。
[0078]
以下通过描述通式i所示化合物的一种典型的合成路线,以进一步描述本发明的技术方案,具体结合以下示出的反应路线可以看出:
[0079]
化合物1与2经缩合反应得到产物3;
[0080]
产物3经碱水解后得到产物4;
[0081]
产物4与化合物经缩合反应得到产物6;
[0082]
产物6经脱保护反应得到产物7;
[0083]
产物7与化合物8经缩合反应得到产物9;
[0084]
产物9经水解反应得到产物10。
[0085][0086]
其中,n=1-3。
[0087]
作为上述典型的反应路线的一种变化,以下示出了另一种反应路线:
[0088]
化合物10经脱保护得到产物11;
[0089]
产物11与化合物12反应后得到产物13。
[0090]
[0091]
其中n为1-3的整数,x-r1,y-r2,z-r3的定义如上所述。
[0092]
下面的实施例可以对本发明做进一步的描述,然而,这些实施例不应作为对本发明的范围的限制。
[0093]
本发明中使用的缩写如下:
[0094]
acn:乙腈
[0095]
cdcl3:氘待三氯甲烷
[0096]
cdi:n,n
’-
羰基二咪唑
[0097]
cd3od:氘代甲醇
[0098]
dipea:n,n
’-
二异丙基乙胺
[0099]
dcm:二氯甲烷
[0100]
dmso:二甲基亚砜
[0101]
dmso-d6:氘待二甲基亚砜
[0102]
edci:1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐
[0103]
esi:电喷雾离子源
[0104]
hcooh:甲酸
[0105]1h-nmr:核磁共振氢谱
[0106]
hplc:高效液相色谱法
[0107]
hobt:1-羟基苯并三唑
[0108]
hz:赫兹
[0109]
h2o:水
[0110]
lc-ms:液相色谱-质谱联用仪
[0111]
lioh:氢氧化锂
[0112]
mhz:兆赫兹
[0113]
ms:质谱
[0114]
nmr:核磁共振
[0115]
petroleum ether:石油醚
[0116]
tea:三乙胺
[0117]
tfa:三氟醋酸
[0118]
thf:四氢呋喃
[0119]
以下描述本发明实施例中通用试验条件:
[0120]
首先,实施例中的反应一般在氮气保护下进行。
[0121]
进一步地,中间体和最终产物通过色谱柱、制备色谱板和icso快速制备色谱系统分离纯化。
[0122]
进一步地,lc-ms液质联用色谱仪使用waters公司acquity arc配备qda detector。质谱(ms)采用esi源,仅指示母体分子的分子量m,通常汇报[m+h]
+
。注射体积是通过样品浓度来确定;流速为:0.8ml/min;hplc的峰值是通过在220nm和254nm处的uv-vis波长来记录读取的。流动相为0.01%甲酸的超纯水溶液(流动相a)和0.01%甲酸的乙腈溶液(流动相b)。梯度洗脱条件如下表1和表2所示:
[0123]
表1:梯度洗脱条件1
[0124]
时间(min)a(h2o,0.01%hcooh)b(ch3cn,0.01%hcooh)0.0-0.395-855-150.3-3.285-2015-803.2-3.820-580-953.8-3.815-9595-53.81-4.0955
[0125]
表2:梯度洗脱条件2
[0126]
时间(min)a(h2o,0.01%hcooh)b(ch3cn,0.01%hcooh)0.00-5.9095-55-955.90-5.915-9595-55.91-6.00955
[0127]
进一步地,nmr谱图采用varian 400mhz核磁共振谱仪获得数据,常以cdcl3,dmso-d6作为溶剂,以ppm报告化学位移。各种峰的描述如下:s(单峰),d(双峰),t(三重峰),q(四重峰),m(多重峰),dd(双二重峰)。偶合常数使用hz表示。
[0128]
实施例1:(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0129][0130]
步骤1a:制备n-叔丁氧羰基-l-缬氨酰-l-脯氨酸甲酯
[0131][0132]
氮气保护下,将n-叔丁氧羰基-l-缬氨酸55.0g、l-脯氨酸甲酯盐酸盐46.1g和n,n'-羰基二咪唑43.1g溶于200ml二氯甲烷中,室温搅拌6小时。hplc监测反应完全。反应液分别用碳酸氢钠溶于、稀盐酸、水洗涤。减压浓缩除去溶剂,得到淡黄色油状物中间体n-叔丁氧羰基-l-缬氨酰-l-脯氨酸甲酯(72.0g)。
[0133]
步骤1b:制备n-叔丁氧羰基-l-缬氨酰-l-脯氨酸
[0134][0135]
氮气保护下,将中间体n-叔丁氧羰基-l-缬氨酰-l-脯氨酸甲酯72.0g、氢氧化锂18.4g溶于360ml四氢呋喃和120ml水中,室温下搅拌3小时。hplc监测反应完全,反应液中加入2%氢氧化钠溶液350ml,反应液用乙酸乙酯洗涤。水相用盐酸调节ph到3~4,然后用乙酸乙酯萃取。减压旋干溶剂,粗品加入10ml乙酸乙酯和400ml正庚烷,室温搅拌5小时,过滤并用50ml淋洗滤饼,减压干燥得到白色固体中间体n-叔丁氧羰基-l-缬氨酰-l-脯氨酸(59.6g)。
[0136]
步骤1c:制备((s)-3-甲基-1((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酸叔丁酯
[0137][0138]
氮气保护下,将中间体n-叔丁氧羰基-l-缬氨酰-l-脯氨酸15.0g、(r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基-1-胺17.8g、1-羟基苯并三唑8.4g溶于300ml二氯甲烷中,室温下缓慢滴加n,n-二异丙基乙胺24.7g,升温至回流并继续搅拌6小时。hplc监测反应完全。反应液用碳酸氢钠溶液、稀盐酸、水洗涤。减压浓缩除去溶剂,得到淡黄色油状物中间体((s)-3-甲基-1((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酸叔丁酯(27.04g)。
[0139]
步骤1d:制备(s)-1-(l-缬氨酰)-n-((r)-2-甲基-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)吡咯烷-2-甲酰胺
[0140][0141]
氮气保护下,将中间体((s)-3-甲基-1((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡
咯烷-1-基)-1-氧代丁-2-基)氨基甲酸叔丁酯34.62g溶于300ml二氯甲烷中,降温至0℃,缓慢加入三氟醋酸60ml,然后继续搅拌6小时。hplc监测反应完全,减压浓缩除去溶剂,然后加入200ml二氯甲烷,用碳酸氢钠溶液调节ph到9,静置分层,水相用二氯甲烷萃取,合并有机相,有机相用水洗,有机相减压浓缩除去溶剂,得到淡黄色油状物中间体(s)-1-(l-缬氨酰)-n-((r)-2-甲基-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)吡咯烷-2-甲酰胺(30.9g)。
[0142]
步骤1e:制备(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸甲酯
[0143][0144]
氮气保护下,将中间体(s)-1-(l-缬氨酰)-n-((r)-2-甲基-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)吡咯烷-2-甲酰胺30.9g、4-((2-甲氧基-2-氧代乙基)氨基甲酰)苯甲酸18.0g溶于300ml二氯甲烷中,室温下缓慢滴加三乙胺31.4g,然后加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐15.8g、1-羟基苯并三唑11.1g,升温回流并继续搅拌6小时。hplc监测反应完全。反应液用碳酸氢钠溶液、稀盐酸、水依次洗涤。减压浓缩除去溶剂。柱层析纯化得到白色固体(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸甲酯(21.1g)。
[0145]
步骤1f:制备(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0146][0147]
氮气保护下,将中间体(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸甲酯2.44g、一水氢氧化锂0.31g溶于30ml四氢呋喃和10ml水中,并在室温搅拌2小时。hplc监测反应完全。浓缩反应
液,然后向浓缩液中加入15ml水,用盐酸调节体系ph=4。溶液用100ml二氯甲烷萃取3次,合并有机相,浓缩得到白色固体粗品。粗品加入2ml乙酸乙酯和10ml正庚烷,室温搅拌5小时,过滤并用5ml淋洗滤饼,减压干燥得到白色固体(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸(2.27g),收率95.0%。1h-nmr(cd3od,400mhz)δ7.89-7.98(m,4h),4.57-4.66(m,2h),4.15-4.21(m,1h),4.02-4.11(m,3h),3.75-3.83(m,1h),2.49(d,j=6.78hz,1h),2.02-2.36(m,7h),1.95(t,j=5.40hz,1h),1.77-1.88(m,3h),1.50(d,j=10.29hz,1h),1.34(s,3h),1.28(s,3h),1.08(dd,j=11.04,6.53hz,6h),0.95-1.02(m,7h),0.85-0.89(m,3h)。ms实测值(esi
+
)[(m+h)
+
]:653。
[0148]
实施例2:(4-(((s)-3-甲基-1-(s)-2-(((r)-2-甲基-1-(6-甲基-4,8-二氧-1,3,6,2-二氧氮杂硼烷-2-基)丙基)氨基甲酰)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0149][0150]
步骤2a:制备(4-(((s)-1-((s)-2-(((r)-二羟硼基-2-甲基丙基)吡咯烷-1-基)-3-甲基-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0151][0152]
氮气保护下,将中间体(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-((3as,4s,6s,7ar)-3a,5,5-三甲基六氢-4,6-甲桥苯并[d][1,3,2]二噁硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸7.4g、4-三氟甲基苯硼酸2.4g溶于100ml乙腈和250ml石油醚中,然后加入10ml浓盐酸,升温至60℃并继续搅拌4小时,静置分层,分出上层的石油醚层,加入250ml新鲜的石油醚并继续在60℃下搅拌4小时。hplc监测反应完全。静置分层,乙腈层用石油醚洗涤。减压浓缩除去溶剂,粗品加入50ml四氢呋喃和300ml乙酸乙酯,室温下搅拌6小时。过滤并用10ml乙酸乙酯洗涤。减压干燥得到白色固体(4-(((s)-1-((s)-2-(((r)-二羟硼基-2-甲基丙基)吡咯烷-1-基)-3-甲基-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸(2.75g),收率50.8%,纯度99.0%。
[0153]
步骤2b:制备(4-(((s)-3-甲基-1-(s)-2-(((r)-2-甲基-1-(6-甲基-4,8-二氧-1,3,6,2-二氧氮杂硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0154][0155]
氮气保护下,将(4-(((s)-1-((s)-2-(((r)-二羟硼基-2-甲基丙基)吡咯烷-1-基)-3-甲基-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸300mg、n-甲基亚胺二乙酸102mg溶于2ml二甲基亚砜中,然后升温至120℃并继续搅拌10小时,tlc监测反应完全。反应液用反相柱纯化(梯度:0.01%甲酸乙腈:0.01%甲酸水,0%~20%),冻干得到白色固体(4-(((s)-3-甲基-1-(s)-2-(((r)-2-甲基-1-(6-甲基-4,8-二氧-1,3,6,2-二氧氮杂硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸(70mg),收率19%。1h nmr(400mhz,cd3od)δ7.94(s,4h),4.65-4.61(m,1h),4.53-4.49(m,1h),4.14-4.01(m,6h),3.84(s,1h),3.78-3.72(m,1h),3.58(dd,j=4.0hz,12.0hz,1h),2.92(s,3h),2.28-2.16(m,2h),2.10-1.99(m,4h),1.12-1.07(m,6h),0.93(dd,j=4.0hz,8.0hz,6h)。ms实测值(esi
+
)[(m+h)
+
]:630。
[0156]
实施例3:(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0157][0158]
步骤3a:制备(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸
[0159][0160]
氮气保护下,将(4-(((s)-1((s)-2-(((r)-二羟硼基-2甲基丙基)吡咯烷-1-基)-3-甲基-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸100mg、频哪醇68mg溶于0.5ml二甲基亚砜中,然后升温至120℃并继续搅拌6小时,lc-ms监测反应完全。反应液冻干后固体用
乙酸乙酯溶解,用制备板纯化,冻干得到白色固体(4-(((s)-3-甲基-1-((s)-2-(((r)-2-甲基-1-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)丙基)氨基甲酰基)吡咯烷-1-基)-1-氧代丁-2-基)氨基甲酰基)苯甲酰基)甘氨酸(45mg),收率39%。1h nmr(400mhz,cd3od)δ7.97-7.90(m,4h),4.67-4.58(m,2h),4.08(br.s.,4h),3.79(d,j=6.44hz,2h),2.41(d,j=6.19hz,1h),2.15-2.36(m,4h),2.01-2.12(m,2h),1.84(td,j=13.21,6.57hz,2h),1.14-1.25(m,12h),1.10(dd,j=15.21,6.70hz,6h),1.01(d,j=6.70hz,2h),0.91-0.98(m,4h)。ms实测值(esi
+
)[(m+h)
+
]:601。
[0161]
稳定性测试
[0162]
根据文献报道配置了gamble’s缓冲液和alf缓冲液两种模拟肺液(marques mrc,loebenberg r,almukainzi m(2011),“simulated biological fluids with possible application in dissolution testing”,dissolution technologies 18(3):15-28)用于测试前药在模拟肺液中稳定性。另外根据动物实验制剂及样品分析的需要,配置了三种不同溶剂:动物实验用溶媒(加5%乙醇的水),无水乙醇,和乙腈,用于测试动物给药前制剂的稳定性和采集的样品在分析过程中的稳定性。分别取实施例1和2的化合物2毫克,溶于1毫升上述过滤过的缓冲液或者溶剂中,漩涡震荡后观测有无未溶的颗粒物,然后将悬浊液或者溶液过滤清除未溶化合物,并立即取100微升,作为稳定性测试开始时间点样品,用hplc方法分析其中前药和水解产物wo2018175173中化合物52的化合物(对照物)的比例。之后两个模拟肺液中的样本放置于37℃定轨摇床200rpm摇晃,并于0.5小时,1小时,2小时,4小时,20小时分别取样并立即用hplc方法测试前药与水解产物比例。对于溶于三个溶剂中的样品,则室温静置,并于1小时,3小时,18小时等时间点分别取样立即检测前药与水解产物比例。
[0163]
检测结果:
[0164]
实施例1和2的化合物显示出截然不同的水解曲线和特征。如图1所示,实施例1化合物在偏中性的gamble’s缓冲液(ph 7.4)和偏酸性的alf缓冲液(ph 4.5)两种模拟肺液中溶解后在半小时内水解分别达到25%和40%,并在其后的20个小时内都维持在这个比例,即达到了稳定平衡状态。如图2所示,实施例2化合物的水解速度为线性,在偏中性的gamble’s缓冲液(ph 7.4)中水解得更快,在2.5小时接近50%水解,而在偏酸性的alf缓冲液(ph 4.5)中水解稍慢,大约15小时水解约50%。如图3所示,实施例1的化合物在乙腈和无水乙醇中几乎不水解;在溶媒中,实施例1的化合物在3小时水解约18%,随后水解速度减慢,在18小时水解约20%。如图4所示,实施例2化合物在三种溶剂中的水解速度测试也显示出线性稳定的水解速度,其中在乙氰中不水解,在无水乙醇中大约每小时有0.5%的化合物水解,在溶媒中则大约为每小时水解1%。
[0165]
体内药代动力学实验
[0166]
实施例1制得的最终前药化合物和对照物(wo2018175173a1中化合物52)在小鼠体内的药代动力学:
[0167]
7-9周大的雄性c57bl/6小鼠经过适应阶段后随机分成两组:弹性蛋白酶抑制剂对照物组和实施例1组,给药细节如表3所示:
[0168]
表3:小鼠体内药代动力学实验设计
[0169]
分组化合物动物数量给药方式剂量
第一组对照物4气管内给药2mg/kg第二组实施例112气管内给药2mg/kg
[0170]
小鼠体内药代动力学研究
[0171]
在小鼠体内药代动力学的实验结果中,对照物在肺部6小时药物浓度为153ng/g,在12小时完全清除,如图5所示。而实施例1组,其为对照物的前药,其在小鼠体内很快转化为对照物。如图6所示,在6小时和12小时,前药实施例1在肺部浓度分别为在1070ng/g和120ng/g,其有抑制活性的水解产物(对照物)在肺部6小时和12小时的浓度也在931ng/g和264ng/g。结果显示,相比直接给与对照物本身,前药实施例1大大提高了对照物在肺部的浓度和停留时间。实施例1化合物相对于对照物在体内pk方面的显著改进。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1