一种2-甲基呋喃和2,5-二甲基呋喃的分离方法与流程
本发明涉及吸附分离技术领域,具体涉及一种2-甲基呋喃和2,5-二甲基呋喃的分离方法。
背景技术:
2-甲基呋喃和2,5-二甲基呋喃的分离是化工和石化行业中具有很大挑战性的任务之一。2-甲基呋喃是一种重要的石油化工产品,可用于合成戊二烯、戊二醇和2-甲基四氢呋喃,在医药工业中,用于合成维生素b1、磷酸氯喹和磷酸伯氨喹,同时也是农药杀虫剂拟除虫菊酯类杀虫剂中间体,是一种具有重要工业和环境意义的挥发性有机化合物。2,5-二甲基呋喃是一种重要的医药中间体,也被认为是可用于替代乙醇燃料的新型生物燃料。在化学工业中,2-甲基呋喃和2,5-二甲基呋喃主要来源于相应的醛醇催化加氢反应。为了同时获得高纯度的2-甲基呋喃和2,5-二甲基呋喃,将两者分别从混合物中选择性分离是至关重要的。然而,由于2-甲基呋喃和2,5-二甲基呋喃具有相似的物理性质,在化工生产中很难分离。由于2-甲基呋喃(336k)和2,5-二甲基呋喃(366k)的沸点比较接近,且共沸物的形成使它们通过传统的蒸馏过程分离出来的纯度不高。目前,分离2-甲基呋喃和2,5-二甲基呋喃混合物的主要工业方法是萃取精馏和共沸精馏。然而,这些方法需要很高的能量,伴随着过程复杂性和高运营成本。因此,开发易于操作和更节能的方法来分离2-甲基呋喃和2,5-二甲基呋喃是必要和可取的。
公开号为cn110054602a的专利说明书公布了一种糠醛加氢制2-甲基呋喃的方法。该发明的目的是要解决现有以cu为活性组分的cu-cr催化剂糠醛加氢生产2-甲基呋喃时存在选择性低的问题。方法:以磷化钴催化剂作为催化剂,催化糠醛加氢脱氧,得到2-甲基呋喃。优点:糠醛的转化率可达到100%,2-甲基呋喃的选择性达到89%以上,且易于实现工业化生产。虽然该发明选择性与之前相比有所提高,但是仍然没有达到95%以上。
公开号为cn110180550a的专利说明书公布了一种糠醛高效转化为2-甲基呋喃催化剂的制备方法,该发明通过沉淀与水热协同作用制备方法实现活性和稀土等改性组分的高分散。催化剂组成为氧化铜含量20-50%,氧化硅含量为40-70%,氧化铝含量为0-8%,稀土氧化物,如la、ce、pr、nd、sc、y等稀土元素中的一种或两种调控改性cu催化剂,稀土氧化物含量为1-6%。该发明合成的绿色环保型催化剂用于糠醛气相加氢制备2-甲基呋喃,糠醛转化率为100%,2-甲基呋喃选择性达90%以上,连续运行500小时性能稳定,但是得到的2-甲基呋喃纯度仍然不高。
公开号为cn109535108a的专利说明书公布了一种2,5-二甲基呋喃的制备方法。该制备方法包括如下步骤:将纤维素、醋酸锰溶液加入氢氧化钠/尿素溶液中,进行碳化处理得到催化剂;该催化剂应用于5-羟甲基糠醛合成2,5-二甲基呋喃反应,包括如下步骤:将5-羟甲基糠醛加入有机醇类溶剂中,制成溶液;将溶液和催化剂混合置于反应釜中,用氮气排出空气,加热搅拌进行加氢脱氧反应,得到2,5-二甲基呋喃。该发明利用催化剂制备2,5-二甲基呋喃的方法虽然易操作,成本低,合成方法较简单,但是分离纯度不高。
公开号为cn109985664a的专利说明书公布了一种用于一步法催化果糖转化为2,5-二甲基呋喃的酸性固体催化剂及其制备方法与应用。该催化剂由负载有金属纳米颗粒的酸性多孔聚合物构成,所述多孔聚合物为多功能聚合物,具有不同浸润性和酸性,可在较温和的条件下锚定金属钌、铂、铑、钯、镍、钴。将该催化剂用于一步催化转化果糖为2,5-二甲基呋喃,催化活性较好、选择性高、稳定性强,但是制备的2,5-二甲基呋喃纯度不高。
利用2-甲基呋喃和2,5-二甲基呋喃在分子尺寸和几何形状上的差异,利用有序多孔材料进行吸附分离是一种有效的分离方法。例如,像金属有机框架材料及共价有机框架可用于吸附分离2-甲基呋喃和2,5-二甲基呋喃。然而,由于它们的分子尺寸非常接近,设计和合成合适的金属有机框架材料用于2-甲基呋喃和2,5-二甲基呋喃的分离是一个挑战。此外,由可逆的金属-配位键构成的金属有机框架材料在实际回收应用中不够稳定。因此,迫切需要开发新型稳定的、可回收的吸附材料来有效分离2-甲基呋喃和2,5-二甲基呋喃。
技术实现要素:
针对本领域存在的不足之处,以及2-甲基呋喃和2,5-二甲基呋喃分离技术中存在的耗能大、过程繁琐、需要使用高纯度脱附剂等缺陷,本发明提供了一种2-甲基呋喃和2,5-二甲基呋喃的分离方法,利用柱[5]芳烃晶体材料和/或杂[3]芳烃晶体材料高选择性吸附分离2-甲基呋喃和2,5-二甲基呋喃混合物,能耗低、过程简单。
一种2-甲基呋喃和2,5-二甲基呋喃的分离方法,利用柱[5]芳烃晶体材料和/或杂[3]芳烃晶体材料吸附分离2-甲基呋喃和2,5-二甲基呋喃混合物;
所述柱[5]芳烃晶体材料的结构如下式(i)所示:
所述杂[3]芳烃晶体材料的结构如下式(ii)所示:
由于2-甲基呋喃和2,5-二甲基呋喃的分子结构的差别,所述柱[5]芳烃晶体材料在常温下能够与2-甲基呋喃形成稳定的化学计量比为1:2的主客体络合物,但是2-甲基呋喃与柱[5]芳烃形成的主客体络合物是在加热时是不稳定的,会逐渐解络合,将吸附的2-甲基呋喃释放出来。与柱[5]芳烃不同的是,杂[3]芳烃晶体材料与2,5-二甲基呋喃形成化学计量比为1:1的主客体络合物。杂[3]芳烃与2,5-二甲基呋喃形成的主客体络合物在常温下同样是稳定的,但在加热时会逐渐解络合,将吸附的2,5-二甲基呋喃释放出来。这体现了两种晶体材料具有不同的选择性。所述柱[5]芳烃和杂[3]芳烃晶体材料在脱附温度下是稳定的,在脱附过程完成后,可以重复利用,且选择性不会下降。
所述柱[5]芳烃和杂[3]芳烃晶体材料为现有材料,前者如erruili等人已在《angewandtechemieinternationaledition》2019年58卷第3981~3985页公开,后者如jiongzhou等人已在《chemicalcommunications》2016年52卷第1622~1624页公开。
作为优选,所述柱[5]芳烃晶体材料、杂[3]芳烃晶体材料通过在不良溶剂中重结晶后活化得到,然后再用于吸附分离2-甲基呋喃和2,5-二甲基呋喃混合物。
所述不良溶剂可以为甲醇,但不限于此。
重结晶得到的柱[5]芳烃晶体材料、杂[3]芳烃晶体材料可以通过加热的方式除去溶剂分子进行活化。作为优选,所述活化的温度不低于150℃,时间不小于4小时。活化后的柱[5]芳烃或杂[3]芳烃晶体材料可以直接用于2-甲基呋喃和2,5-二甲基呋喃混合物的选择性吸附分离。
作为优选,所述利用柱[5]芳烃晶体材料和/或杂[3]芳烃晶体材料吸附分离2-甲基呋喃和2,5-二甲基呋喃混合物的具体步骤为:将所述柱[5]芳烃晶体材料和/或杂[3]芳烃晶体材料置于2-甲基呋喃和2,5-二甲基呋喃的混合蒸气氛围内,温度小于60℃。吸附时间可随样品量和2-甲基呋喃或者2,5-二甲基呋喃在混合物中的比例等因素的改变而改变。在吸附过程中,所述柱[5]芳烃晶体材料、杂[3]芳烃晶体材料会发生晶型的改变。由于ch-π、ch-o之间的多重非共价键相互作用,混合蒸气中的2-甲基呋喃、2,5-二甲基呋喃会分别与柱[5]芳烃、杂[3]芳烃形成主客体络合物,该主客体络合物的化学计量比分别是1:2或者1:1。
吸附完毕后,取出所述柱[5]芳烃、杂[3]芳烃晶体材料,然后可采用真空加热或减压加热除去柱[5]芳烃晶体材料和/或杂[3]芳烃晶体材料表面吸附的2-甲基呋喃和2,5-二甲基呋喃混合物。作为优选,所述真空加热或减压加热的温度小于60℃。加热时间可根据样品量进行调整。在低于60℃的条件下所述主客体络合物依然稳定存在,而表面吸附的2-甲基呋喃和2,5-二甲基呋喃混合物则可以逐渐除去。通过除去表面吸附的混合物,吸附分离出的客体纯度可进一步提高。
可采用加热脱附的方式除去柱[5]芳烃、杂[3]芳烃晶体材料吸附络合的客体,同时实现柱[5]芳烃、杂[3]芳烃晶体材料的再生。脱附时间可随样品量进行调整。
作为优选,加热至60~100℃脱附柱[5]芳烃晶体材料吸附络合的2-甲基呋喃,实现柱[5]芳烃晶体材料的再生。
作为优选,加热至60~100℃脱附杂[3]芳烃晶体材料吸附络合的2,5-二甲基呋喃,实现杂[3]芳烃晶体材料的再生。
在上述温度下,主客体络合物是不稳定的,被吸附的客体分子会逐渐释放出来,而柱[5]芳烃、杂[3]芳烃晶体材料则是稳定的,在脱附的过程中只是发生晶型的改变。脱附完成后即得到再生的柱[5]芳烃、杂[3]芳烃晶体材料,可以继续用于吸附分离2-甲基呋喃或者2,5-二甲基呋喃,进行下一次循环。
本发明还提供了所述的如式(i)结构所示的柱[5]芳烃晶体材料在选择性吸附络合2-甲基呋喃中的应用。
本发明还提供了所述的如式(ii)结构所示的杂[3]芳烃晶体材料在选择性吸附络合2,5-二甲基呋喃中的应用。
本发明与现有技术相比,主要优点包括:分离过程操作简单,设备要求低;分离过程不需要精馏操作,能耗低,节约能源,降低了2-甲基呋喃和2,5-二甲基呋喃的生产成本;所用晶体材料稳定性高,可以循环使用,分离效果不会降低。
附图说明
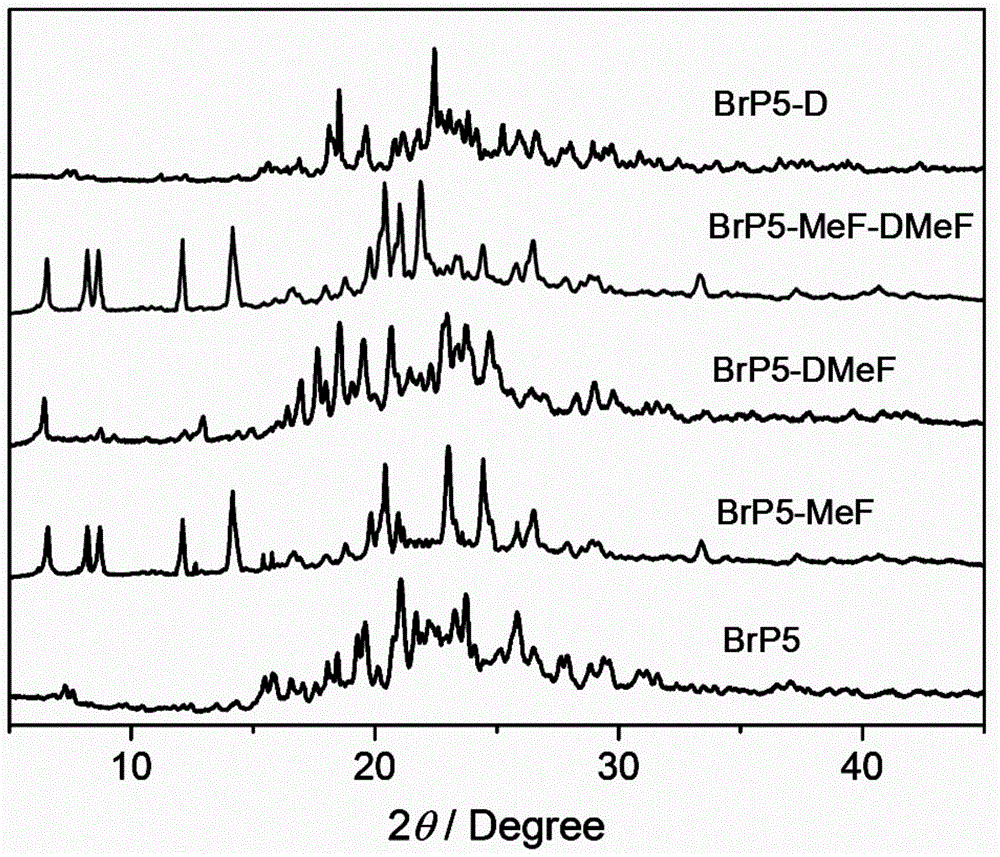
图1为实施例1~4的柱[5]芳烃晶体材料的粉末x射线衍射(pxrd)图;
图2为实施例3的柱[5]芳烃晶体材料吸附分离2-甲基呋喃和2,5-二甲基呋喃的气相色谱表征结果图,横坐标代表时间,单位min;
图3为实施例5的柱[5]芳烃晶体材料循环使用时对2-甲基呋喃和2,5-二甲基呋喃吸附分离效果图;
图4为实施例6~9的杂[3]芳烃晶体材料的粉末x射线衍射(pxrd)图;
图5为实施例8的杂[3]芳烃晶体材料吸附分离2-甲基呋喃和2,5-二甲基呋喃的气相色谱表征结果图,横坐标代表时间,单位min;
图6为实施例10的杂[3]芳烃晶体材料循环使用时对2-甲基呋喃和2,5-二甲基呋喃吸附分离效果图。
具体实施方式
下面结合附图及具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的操作方法,通常按照常规条件,或按照制造厂商所建议的条件。
实施例1
柱[5]芳烃晶体材料的制备:称取4g柱[5]芳烃置于20ml甲醇中,加热至沸腾,滴加甲醇直至全部溶解,将溶液放置于0℃下保存过夜,过滤收集析出来的晶体,将得到的晶体于60℃真空干燥,在150℃下活化4小时,得到白色粉末,记为brp5。
本实施例制备的产品表征数据如下:
brp5,1hnmr(400mhz,chloroform-d,293k,ppm)δ6.91(s,10h),4.21-4.24(t,20h),3.84(s,10h),3.61-3.64(t,20h)。
pxrd检测结果如图1所示,所得到的柱[5]芳烃晶体材料具有良好的结晶度。
实施例2
柱[5]芳烃晶体材料对单独2-甲基呋喃和2,5-二甲基呋喃的吸附:取两个20ml菌种瓶,分别加入1ml2-甲基呋喃和1ml2,5-二甲基呋喃,命名为brp5-mef和brp5-dmef,分别取400mg实施例1制得的柱[5]芳烃晶体材料放置于两个5ml敞口菌种瓶中,将两个敞口5ml菌种瓶分别置于两个20ml菌种瓶中,将20ml菌种瓶密封好,置于25℃水浴锅中30小时。
本实施例制备的产品表征数据如下:
brp5-mef,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.28(s,2h),6.91(s,10h),6.27(s,2h),5.96(s,2h),4.21-4.24(t,20h),3.84(s,10h),3.61-3.64(s,20h),2.29(s,6h)。
brp5-dmef,1hnmr(400mhz,chloroform-d,293k,ppm)δ6.91(s,10h),5.83(s,3h),4.21-4.24(t,20h),3.84(s,10h),3.61-3.64(s,20h),2.24(s,9h)。
1hnmr结果表明柱[5]芳烃晶体材料以化学计量比为1:2的方式吸附了2-甲基呋喃,对2,5-二甲基呋喃则吸附的不多。
pxrd检测结果如图1所示,相对于最初活化的柱[5]芳烃晶体材料的pxrd谱图,在2-甲基呋喃蒸气中放置了一段时间后的柱[5]芳烃晶体材料的pxrd谱图出现变化,这说明它的晶胞参数已经发生了变化,意味着2-甲基呋喃已经被吸附进入柱[5]芳烃晶体材料;在2,5-二甲基呋喃蒸气中放置了一段时间后的柱[5]芳烃晶体材料的谱图变化不大,说明它的晶胞参数几乎没有变化,意味着柱[5]芳烃晶体材料对2,5-二甲基呋喃吸附能力不强。
实施例3
柱[5]芳烃晶体材料对2-甲基呋喃和2,5-二甲基呋喃的1:1混合物的吸附:取一个20ml菌种瓶,加入0.5ml2-甲基呋喃和0.5ml2,5-二甲基呋喃,命名为brp5-mef-dmef,取400mg实施例1制得的柱[5]芳烃晶体材料放置于5ml敞口菌种瓶中,将敞口5ml菌种瓶置于上述20ml菌种瓶中,将20ml菌种瓶密封好,置于25℃水浴锅中40小时,将得到的粉末在50℃真空烘箱中放置30分钟。
本实施例制备的产品表征数据如下:
brp5-mef-dmef,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.28(s,2h),6.91(s,10h),6.27(s,2h),5.96(s,2h),4.21-4.23(t,20h),3.84(s,10h),3.61-3.63(s,20h),2.29(s,6h)。
在1hnmr谱图中发现了2-甲基呋喃所对应的氢原子的信号,2,5-二甲基呋喃对应的信号几乎可以忽略,这说明柱[5]芳烃晶体材料可以选择性地吸附2-甲基呋喃。
pxrd检测结果如图1所示,相对于最初活化的柱[5]芳烃晶体材料的pxrd谱图,在2-甲基呋喃和2,5-二甲基呋喃的混合蒸气中放置了一段时间后的柱[5]芳烃晶体材料的pxrd谱图出现变化,并且谱图变化与brp5-mef相同,这说明柱[5]芳烃晶体材料可以选择性的吸附2-甲基呋喃。
顶空气相色谱的结果如图2所示,结果表明,柱[5]芳烃晶体材料可以选择性的吸附2-甲基呋喃,其选择性为96.3%。
实施例4
柱[5]芳烃晶体材料再生:将饱和吸附2-甲基呋喃的柱[5]芳烃晶体材料400mg在真空烘箱100℃下加热4小时,样品记为brp5-d。
本实施例制备的产品表征数据如下:
brp5-d,1hnmr(400mhz,chloroform-d,293k,ppm)δ6.91(s,10h),4.21-4.24(t,20h),3.84(s,10h),3.61-3.64(t,20h)。
在1hnmr谱图中发现2-甲基呋喃所对应的氢原子的信号已经消失,这说明柱[5]芳烃晶体材料已经完成了脱附再生,2-甲基呋喃分子已经全部释放。
pxrd检测结果如图1所示,相对于最初活化的柱[5]芳烃晶体材料的pxrd谱图,脱附完全后的柱[5]芳烃晶体材料的pxrd谱图变化不大,这说明柱[5]芳烃晶体材料已经完成脱附过程,可用于下一次的吸附分离2-甲基呋喃和2,5-二甲基呋喃。
实施例5
柱[5]芳烃晶体材料重复利用:将再生后的柱[5]芳烃晶体材料400mg重复实施例3、4。
顶空气相色谱的结果如图3所示,说明柱[5]芳烃晶体材料可以选择性的吸附2-甲基呋喃,其选择性高达96.3%,而且在重复使用5次其选择性没有降低。
实施例6
杂[3]芳烃晶体材料的制备:称取2g杂[3]芳烃置于20ml甲醇中,加热至沸腾,滴加甲醇直至全部溶解,将溶液放置于0℃下保存过夜,过滤收集析出来的晶体,将得到的晶体于50℃真空干燥,在150℃下活化4小时,得到白色粉末,记为1。
本实施例制备的产品表征数据如下:
1,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.16-7.18(d,j=8hz,2h),6.72-6.74(d,j=8hz,2h),6.35(s,2h),6.29(s,2h),4.05-4.19(m,8h),3.89(s,6h),3.71(s,6h),3.63(s,6h),3.45-3.49(d,j=16hz,2h),1.43-1.47(t,j=8hz,6h)。
pxrd检测结果如图4所示,所得到的杂[3]芳烃晶体材料具有良好的结晶度。
实施例7
杂[3]芳烃晶体材料对单独2-甲基呋喃或2,5-二甲基呋喃的吸附:取两个20ml菌种瓶,分别加入1ml2-甲基呋喃和1ml2,5-二甲基呋喃,命名为1-mef和1-dmef,分别取200mg实施例6制得的杂[3]芳烃晶体材料放置于两个5ml敞口菌种瓶中,将两个敞口5ml菌种瓶分别置于两个20ml菌种瓶中,将20ml菌种瓶密封好,置于25℃水浴锅中30小时。
本实施例制备的产品表征数据如下:
1-mef,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.16-7.18(d,j=8hz,2h),6.72-6.74(d,j=8hz,2h),6.35(s,2h),6.29(s,2h),4.05-4.19(m,8h),3.89(s,6h),3.71(s,6h),3.63(s,6h),3.45-3.49(d,j=16hz,2h),1.43-1.47(t,j=8hz,6h)。
1-dmef,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.16-7.18(d,j=8hz,2h),6.72-6.74(d,j=8hz,2h),6.35(s,2h),6.29(s,2h),5.83(s,2h),4.05-4.19(m,8h),3.89(s,6h),3.71(s,6h),3.63(s,6h),3.45-3.49(d,j=16hz,2h),2.25(s,6h),1.43-1.47(t,j=8hz,6h)。
1hnmr结果表明杂[3]芳烃晶体材料以化学计量比为1:1的方式吸附了2,5-二甲基呋喃,对2-甲基呋喃则没有吸附。
pxrd检测结果如图4所示,相对于最初活化的杂[3]芳烃晶体材料的pxrd谱图,在2,5-二甲基呋喃蒸气中放置了一段时间后的杂[3]芳烃晶体材料的pxrd谱图出现变化,这说明它的晶胞参数已经发生了变化,意味着2,5-二甲基呋喃已经被吸附进入杂[3]芳烃晶体材料;在2-甲基呋喃蒸气中放置了一段时间后的杂[3]芳烃晶体材料的谱图变化很小,说明它的晶胞参数几乎没有变化,意味着杂[3]芳烃晶体材料对2-甲基呋喃几乎没有吸附能力。
实施例8
杂[3]芳烃晶体材料2-甲基呋喃和2,5-二甲基呋喃的1:1混合物的吸附:取一个20ml菌种瓶,加入0.5ml2-甲基呋喃和0.5ml2,5-二甲基呋喃,命名为1-mef-dmef,取200mg实施例1制得的杂[3]芳烃晶体材料放置于5ml敞口菌种瓶中,将敞口5ml菌种瓶置于上述20ml菌种瓶中,将20ml菌种瓶密封好,置于25℃水浴锅中40小时,将得到的粉末在60℃真空烘箱中放置30分钟。
本实施例制备的产品表征数据如下:
1-mef-dmef,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.16-7.19(d,j=8hz,2h),6.72-6.75(d,j=8hz,2h),6.35(s,2h),6.29(s,2h),5.83(s,2h),4.05-4.19(m,8h),3.89(s,6h),3.71(s,6h),3.63(s,6h),3.45-3.49(d,j=16hz,2h),2.25(s,6h),1.43-1.47(t,j=8hz,6h)。
在1hnmr谱图中只发现了2,5-二甲基呋喃所对应的氢原子的信号,这说明杂[3]芳烃晶体材料可以选择性地吸附2,5-二甲基呋喃。
pxrd检测结果如图4所示,相对于最初活化的杂[3]芳烃晶体材料的pxrd谱图,在2-甲基呋喃和2,5-二甲基呋喃的混合蒸气中放置了一段时间后的杂[3]芳烃晶体材料的pxrd谱图出现变化,并且谱图变化与1-dmef相同,这说明杂[3]芳烃晶体材料可以选择性的吸附2,5-二甲基呋喃。
顶空气相色谱的结果如图5所示,结果表明,杂[3]芳烃晶体材料可以选择性的吸附2,5-二甲基呋喃,其选择性为98.7%。
实施例9
杂[3]芳烃晶体材料再生:将饱和吸附2,5-二甲基呋喃的杂[3]芳烃晶体材料200mg在真空烘箱100℃下加热4小时,样品记为1-d。
本实施例制备的产品表征数据如下:
1-d,1hnmr(400mhz,chloroform-d,293k,ppm)δ7.16-7.18(d,j=8hz,2h),6.72-6.74(d,j=8hz,2h),6.35(s,2h),6.29(s,2h),4.05-4.19(m,8h),3.89(s,6h),3.71(s,6h),3.63(s,6h),3.45-3.49(d,j=16hz,2h),1.43-1.47(t,j=8hz,6h)。
在1hnmr谱图中发现2,5-二甲基呋喃所对应的氢原子的信号已经消失,这说明杂[3]芳烃晶体材料已经完成了脱附再生,2,5-二甲基呋喃分子已经全部释放。
pxrd检测结果如图4所示,相对于最初活化的杂[3]芳烃晶体材料的pxrd谱图,脱附完全后的杂[3]芳烃晶体材料的pxrd谱图变化很小,这说明杂[3]芳烃晶体材料已经完成脱附过程,可用于下一次的吸附分离2-甲基呋喃和2,5-二甲基呋喃。
实施例10
杂[3]芳烃晶体材料重复利用:将再生后的杂[3]芳烃晶体材料200mg重复实施例8、9。
顶空气相色谱的结果如图6所示,说明杂[3]芳烃晶体材料可以选择性的吸附2,5-二甲基呋喃,其选择性高达98.7%,而且在重复使用5次其选择性没有降低。
此外应理解,在阅读了本发明的上述描述内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!