一种功能化高分子聚合物及其制备方法与流程
本发明属于高分子合成领域,具体涉及一种功能化高分子聚合物及其制备方法。
背景技术:
随着全球塑料生产量的增长,产量可达数百万吨/年。然而废弃塑料的处理主要是焚烧、填埋等,造成了大量的环境污染,因此需要开发新型的材料解决问题。聚六氢-(3,4)-反式苯并呋喃酮、聚丁内酯等能够完全降解至原料单体,具有循环利用的特性。
共聚方法是材料改性的一种重要的合成方法。但是六氢-(3,4)-反式苯并呋喃酮或丁内酯衍生物与其他内酯单体存在着极大的热力学差异,因此六氢-(3,4)-反式苯并呋喃酮或丁内酯衍生物的共聚存在着严峻的挑战。其中,六氢-(3,4)-反式苯并呋喃酮的共聚反应仍无相关报道。因此,实现六氢-(3,4)-反式苯并呋喃酮或丁内酯衍生物的共聚反应具有重要的研究价值。同时共聚物的末端功能化能够赋予材料更多的功能,拓展了材料的应用范围。
技术实现要素:
发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种功能化高分子聚合物。
本发明还要解决的技术问题是提供上述功能化高分子聚合物的制备方法,以丰富聚(γ-丁内酯)、聚六氢-(3,4)-反式苯并呋喃酮的合成方法,填补功能化及共聚体系方面的空白等问题。
为了解决上述技术问题,本发明公开了一种功能化高分子聚合物,其是由第一单体和第二单体制得的聚合物;
其中,所述的第一单体为式ⅰ所述结构式中的任意一种;
其中,第二单体为式ⅱ所述结构式中的任意一种,或不同于第一单体的式ⅰ所述结构式中的任意一种;
其中,所述的功能化高分子聚合物具有如式ⅲ所示的重复单元结构;
其中,p选自1或2;n、m、x、y分别独立地选自10~100中的任意一个整数;
其中,r1选自式ⅳ所示结构中的任意一种;r2、r3独立地选自氢、甲基、亚甲基或环己基;r2、r3不同;优选地,r2选自氢或环己基;更优选地,r2选自环己基;
其中,所述的功能化高分子聚合物的数均分子量为1000~50000gmol-1;
其中,第一单体与第二单体的摩尔比为1~100:1~100。
其中,所述的功能化指的是相对普通的聚合物,本发明申请中的聚合物,其末端r1具有特定的官能团,具有广泛的后修饰功能,如双键、三键可以进行点击化学反应制备星型、网状等复杂构型的聚合物,从而获得不同性质的聚合物;呋喃环能够进行diels-alder反应,属于一种可逆反应,可以制备响应性聚合物;卤素原子能够耦合原子转移自由基聚合,实现两种不同聚合机理反应的结合;聚乙二醇基团具有疏水性,能够获得两亲性聚合物,实现自组装,在药物载体等生物医药方面具有潜在的应用价值。
优选地,所述的第一单体为γ-丁内酯、α-甲基-γ-丁内酯、六氢-(3,4)-反式苯并呋喃酮、(s)-六氢-(3,4)-反式苯并呋喃酮、(r)-六氢-(3,4)-反式苯并呋喃酮;更优选的,所述的第一单体为六氢-(3,4)-反式苯并呋喃酮、(s)-六氢-(3,4)-反式苯并呋喃酮和(r)-六氢-(3,4)-反式苯并呋喃酮中的任意一种;与γ-丁内酯相以及其他内酯单体相比,六氢-(3,4)-反式苯并呋喃酮以及(s)-六氢-(3,4)-反式苯并呋喃酮、(r)-六氢-(3,4)-反式苯并呋喃酮具有明显的优势,其能够在室温条件下发生聚合,而其他单体需要在超低温条件才能反应;此外,六氢-(3,4)-反式苯并呋喃酮以及(s)-六氢-(3,4)-反式苯并呋喃酮、(r)-六氢-(3,4)-反式苯并呋喃酮所制备得到聚合物的热学性能更优。
其中,所述的聚合物具有广泛的热学性能,能够满足不同领域的需求,其熔点为30~150℃;初始降解温度为150~300℃;玻璃化转变温度为-60~70℃。
其中,所述的聚合物根据其制备方法的不同,可分为嵌段共聚物和无规共聚物,两者相比,其结构式相同,性能相似,制备方法不同。
上述功能化高分子聚合物的制备方法也在本发明的保护范围之内。
其中,所述嵌段共聚物的制备方法包括如下步骤:
(1)将第一单体与引发剂混合后,再加入有机镁催化剂,反应,得到反应液;
(2)向步骤(1)得到的反应液中加入溶剂溶解混合物,再加入第二单体,反应,即得;其中,所述的第二单体为式ⅱ所示结构中的任意一种。
其中,所述无规共聚物的制备方法包括如下步骤:
(i)将第一单体、第二单体和引发剂混合;
(ii)向步骤(i)得到的混合溶液中加入有机镁催化剂,反应,即得。
优选地,第二单体为不同于第一单体的式ⅰ所述结构式中的任意一种。
其中,所述嵌段共聚物和无规共聚物制备过程中的引发剂为多官能团脂肪醇和多官能团芳香醇,具体为式ⅴ所述结构式中的任意一种;
其中,所述嵌段共聚物和无规共聚物制备过程中的有机镁催化剂为式ⅵ所述结构式中的任意一种;
其中,y1和y2分别独立选自甲基、乙基、正丁基或异丁基。
其中,所述嵌段共聚物和无规共聚物制备过程中,所述的第一单体、第二单体、引发剂、有机镁催化剂的摩尔比为10~100:10~100:1:0.1~10。
步骤(1)中,所述的反应为在-60~100℃下反应5~1440min。
步骤(2)中,所述的溶剂为二氯甲烷、四氢呋喃、甲苯和乙腈中的任意一种或几种组合;控制溶剂用量使第一单体的浓度4~10mol/l。
步骤(2)中,所述的反应为在-60~100℃下反应1-240min。
步骤(ii)中,所述的反应为在-60~100℃下反应5~1440min。
其中,在嵌段共聚物以及无规共聚物的制备过程中,在步骤(1)、步骤(i)和步骤(b)中可以添加一定的溶剂,所述的溶剂为二氯甲烷、四氢呋喃、甲苯和乙腈中的任意一种或几种组合,若添加溶剂,则控制溶剂用量使第一单体的浓度4~10mol/l。
上述两种不同第一单体制备得到的无规共聚物在制备可化学循环材料中的应用也在本发明的保护范围之内。其中,所述的可化学循环材料为塑料用品,如包装袋、塑料瓶等。
其中,上述两种不同的第一单体制备得到的无规共聚物在一定条件下能够快速降解至单体,并且纯度为99%,不含其他副产物;降解后的产物(无需提纯)可直接重新聚合,获得功能化高分子材料。
其中,降解条件为:催化剂为zncl2(均聚物的1~2mol%),反应温度为60~120℃,甲苯为溶剂(聚合物浓度为0.5g/ml),反应时间为2~12h,回收率为99%;
其中,重新聚合条件:将降解后的产物除去甲苯溶剂,加入有机镁催化剂(有机镁催化剂的摩尔量为引发剂的0.1~10倍),在-60~100℃下反应5~1440min,重新聚合;其中,单体转化率为80~89%。
其中,重新聚合过程中可以添加溶剂,也可不添加溶剂,若要添加溶剂,则加入溶剂(甲苯、四氢呋喃、二氯甲烷或乙腈为溶剂(单体浓度4~10mol/l))。
其中,在重新聚合时,由于催化剂会在空气中失去活性;降解后,引发剂还原至最初结构,因此,催化剂和引发剂不需要去除。
因此,上述两种不同的第一单体制备得到的无规共聚物能够完全降解至单体,无需进一步提纯,降解获得的单体通过重新聚合,可制备功能化聚合物,实现了单体-聚合物-单体的化学循环。
其中,上述反应所涉及的浓度,在计算时以整个体系中液体的体积为溶剂的体积,包括所添加溶剂的体积以及第一单体和第二单体本身,以及部分引发剂和机镁催化剂中的液体。
有益效果:与现有技术相比,本发明具有如下优势:
(1)本发明操作简单,催化剂为商业化化合物,来源广泛,成本低廉,催化活性高且可控性强,转化率高。
(2)本发明的共聚物,比均聚物的热学性能得到了提高。例如,嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的初始分解温度为212℃,最大分解温度为287℃和350℃,差示扫描量热第二次扫描显示熔点为134℃和143℃,玻璃化温度为44℃;均聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮的初始分解温度为196℃,最大分解温度为241℃,差示扫描量热第二次扫描显示无熔点,玻璃化温度为41℃。其热学性能得到了明显的改善。通过该共聚方法,可调控材料的热学性能。
(3)与γ-丁内酯或其他内酯相比,六氢-(3,4)-反式苯并呋喃酮以及(s)-六氢-(3,4)-反式苯并呋喃酮、(r)-六氢-(3,4)-反式苯并呋喃酮具有明显的优势,其能够在室温条件下发生聚合,而其他内酯需要在超低温条件才能反应。同时,六氢-(3,4)-反式苯并呋喃酮以及(s)-六氢-(3,4)-反式苯并呋喃酮、(r)-六氢-(3,4)-反式苯并呋喃酮的热学性能更优,如嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的初始分解温度为212℃,最大分解温度为287℃和350℃,差示扫描量热第二次扫描显示熔点为134℃和143℃,玻璃化温度为44℃;而嵌段共聚物聚γ-丁内酯-b-聚丙交酯的初始分解温度为162℃,最大分解温度为201℃,差示扫描量热第二次扫描显示熔点为88℃,玻璃化温度为-36℃。嵌段共聚物聚α-甲基γ-丁内酯-b-聚戊内酯的初始分解温度为170℃,最大分解温度为211℃,差示扫描量热第二次扫描显示熔点为90℃,玻璃化温度为-30℃。无规共聚物聚α-甲基γ-丁内酯-r-聚丙交酯的初始分解温度为182℃,最大分解温度为240℃,差示扫描量热第二次扫描显示熔点为53℃,玻璃化温度为-56℃
附图说明
图1为嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的热重分析曲线图;
图2为嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的相对导数热重图;
图3为嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的差示扫描量热图;
图4为无规共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚己内酯的热重分析曲线图;
图5为无规共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚己内酯的相对导数热重图;
图6为无规共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚己内酯的差示扫描量热图;
图7为无规共聚物聚(s)-六氢-(3,4)-反式苯并呋喃酮-r-聚三亚甲基碳酸酯的热重分析曲线图;
图8为无规共聚物聚(s)-六氢-(3,4)-反式苯并呋喃酮-r-聚三亚甲基碳酸酯的相对导数热重图;
图9为无规共聚物聚(s)-六氢-(3,4)-反式苯并呋喃酮-r-聚三亚甲基碳酸酯的差示扫描量热图。
图10为嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的体积排阻色谱图。
图11为无规共聚物聚(s)-六氢-(3,4)-反式苯并呋喃酮-r-聚三亚甲基碳酸酯的体积排阻色谱图。
图12为无规共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚己内酯的体积排阻色谱图。
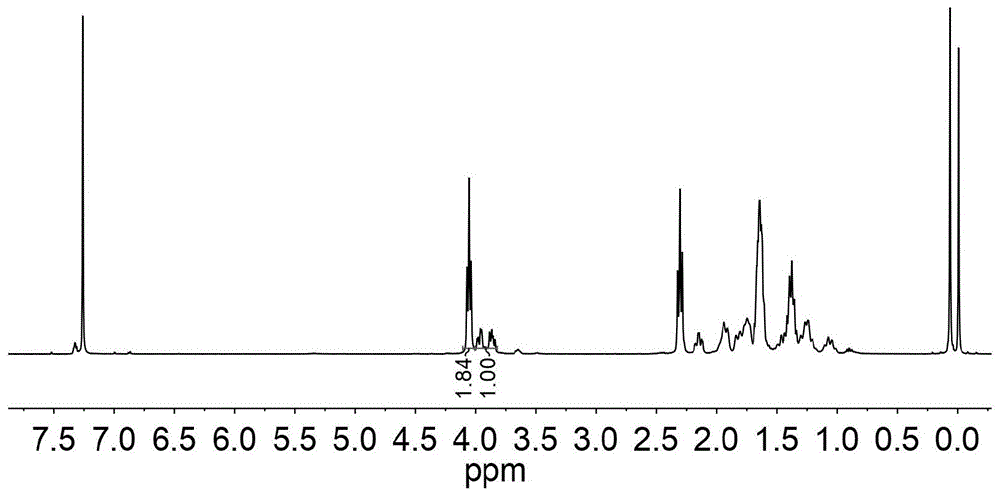
图13为嵌段共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-b-聚丙交酯的核磁共振氢谱图。
图14为无规共聚物聚(s)-六氢-(3,4)-反式苯并呋喃酮-r-聚三亚甲基碳酸酯的核磁共振氢谱图。
图15为无规共聚物聚(r)-六氢-(3,4)-反式苯并呋喃酮-r-聚己内酯的核磁共振氢谱图。
具体实施方式
根据下述实施例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。
以下实施例中所用的二正丁基镁、乙基正丁基镁和正丁基异丁基镁为庚烷溶液1mol/l。
实施例1
将二苯基甲醇(0.0066g,0.036毫摩尔)和(r)-六氢-(3,4)-反式苯并呋喃酮(0.2523g,1.8毫摩尔)加入到无水无氧处理过的安瓿瓶中,200rpm搅拌10分钟之后,加入二正丁基镁(0.06毫升,0.06毫摩尔),置于室温下反应14小时。之后加入无水四氢呋喃0.2ml溶解混合物,然后加入丙交酯(0.2594g,1.8毫摩尔)室温下继续反应20分钟,反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液也1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。丙交酯转化率为99%,(r)-六氢-(3,4)-反式苯并呋喃酮转化率为82%,聚合物数均分子量为7430克/摩尔,分散系数为1.14。如图1、图2、图3、图10和图13所示,其初始降解温度为212℃,最大降解温度为287℃和350℃,熔点为134℃,玻璃化转变温度为44℃。
实施例2
将二苯基甲醇(0.0066g,0.036毫摩尔),三亚甲基碳酸酯(0.1838g,1.8毫摩尔)和(s)-六氢-(3,4)-反式苯并呋喃酮(0.2523g,1.8毫摩尔)加入到无水无氧处理过的安瓿瓶中,300rpm搅拌10分钟之后,加入乙基正丁基镁(0.06毫升,0.06毫摩尔),置于室温下反应12小时。反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。三亚甲基碳酸酯转化率为99%,(s)-六氢-(3,4)-反式苯并呋喃酮转化率为84%,聚合物数均分子量为7600克/摩尔,分散系数为1.27。如图4、图5、图6、图12和图14所示,其初始降解温度为214℃,最大降解温度为265℃,玻璃化转变温度为-20℃。
实施例3
将二苯基甲醇(0.0066g,0.036毫摩尔),己内酯(0.2054g,1.8毫摩尔)和(r)-六氢-(3,4)-反式苯并呋喃酮(0.2523g,1.8毫摩尔)加入到无水无氧处理过的安瓿瓶中,400rpm搅拌10分钟之后,加入二正丁基镁(0.06毫升,0.06毫摩尔),置于室温下反应10小时。反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。己内酯转化率为99%,(r)-六氢-(3,4)-反式苯并呋喃酮转化率为80%,聚合物数均分子量为11320克/摩尔,分散系数为1.44。如图7、图8、图9、图11和图15,其所示初始降解温度为197℃,最大降解温度为290℃,玻璃化转变温度为-40℃。
实施例4
将5-己烯-1-醇(0.0060g,0.06毫摩尔)和γ-丁内酯(0.2583g,3毫摩尔)加入到无水无氧处理过的安瓿瓶中,加入四氢呋喃(0.23ml),300rpm搅拌10分钟之后,加入正丁基异丁基镁(0.06毫升,0.06毫摩尔),置于-60℃反应24小时。之后加入无水四氢呋喃0.3ml溶解混合物,然后加入丙交酯(0.4324g,3毫摩尔)室温下继续反应50分钟,反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液也1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。丙交酯转化率为99%,γ-丁内酯转化率为78%,聚合物数均分子量为10430克/摩尔,分散系数为1.22。其初始降解温度为162℃,最大降解温度为201℃,熔点为88℃,玻璃化转变温度为-36℃。
实施例5
将3-丁炔-1-醇(0.0042g,0.06毫摩尔)α-甲基-γ-丁内酯(0.3004g,3毫摩尔)加入到无水无氧处理过的安瓿瓶中,加入甲苯(0.23ml),260rpm搅拌10分钟之后,加入正丁基异丁基镁(0.06毫升,0.06毫摩尔),置于-40℃反应24小时。之后加入甲苯0.1ml溶解混合物,然后加入戊内酯(0.3004g,3毫摩尔)室温下继续反应30分钟,反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液也1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。戊内酯转化率为99%,α-甲基-γ-丁内酯转化率为78%,聚合物数均分子量为9640克/摩尔,分散系数为1.20。其初始降解温度为170℃,最大降解温度为211℃,熔点为90℃,玻璃化转变温度为-30℃。
实施例6
将苯甲醇(0.0065g,0.06毫摩尔)α-亚甲基-γ-丁内酯(0.2943g,3毫摩尔)、己内酯(0.3424g,3毫摩尔)和甲苯(0.25ml)加入到无水无氧处理过的安瓿瓶中400rpm搅拌10分钟之后,加入二正丁基镁(0.06毫升,0.06毫摩尔),置于-60oc反应12小时。反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。己内酯转化率为92%,(α-亚甲基-γ-丁内酯为82%,聚合物数均分子量为9520克/摩尔,分散系数为1.22。初始降解温度为182℃,最大降解温度为240℃,玻璃化转变温度为-56℃,熔点为53℃。
实施例7
将二苯基甲醇(0.0066g,0.036毫摩尔)和六氢-(3,4)-反式苯并呋喃酮(0.2523g,1.8毫摩尔)加入到无水无氧处理过的安瓿瓶中,200rpm搅拌10分钟之后,加入二正丁基镁(0.06毫升,0.06毫摩尔),置于室温下反应18小时。之后加入无水四氢呋喃0.2ml溶解混合物,然后加入丙交酯(0.2594g,1.8毫摩尔)室温下继续反应20分钟,反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液也1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。丙交酯转化率为99%,六氢-(3,4)-反式苯并呋喃酮转化率为80%,聚合物数均分子量为9660克/摩尔,分散系数为1.18。初始降解温度为223℃,最大降解温度为289℃和352℃,熔点为136℃,玻璃化转变温度为44℃。
实施例8
将苯甲醇(0.0065g,0.06毫摩尔)γ-丁内酯(0.2583g,3毫摩尔)、(r)-六氢-(3,4)-反式苯并呋喃酮(0.4205g,3毫摩尔)和甲苯(0.25ml)加入到无水无氧处理过的安瓿瓶中400rpm搅拌10分钟之后,加入二正丁基镁(0.06毫升,0.06毫摩尔),置于-60℃反应24小时。反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液1hnmr计算得到,聚合物结构通过1hnmr鉴定,聚合物的分子量及分散度通过gpc测定。γ-丁内酯转化率为80%,(r)-六氢-(3,4)-反式苯并呋喃酮的转化率为81%,聚合物数均分子量为9230克/摩尔,分散系数为1.20。初始降解温度为190℃,最大降解温度为245℃,玻璃化转变温度为22℃,熔点为76℃。
实施例9:高分子材料的化学循环:
功能化高分子材料的降解:将实施例8所制备的5-己烯-1-醇末端功能化的聚((r)-六氢-(3,4)-反式苯并呋喃酮)-r-聚(γ-丁内酯)(0.5000克,0.054毫摩尔),zncl2(2mol%),甲苯(1毫升)转移至密闭安瓿瓶中,120℃加热12小时后,产率通过反应液1hnmr计算得到,结构通过1hnmr鉴定。产物为(r)-六氢-(3,4)-反式苯并呋喃酮和γ-丁内酯,转化率为99%。
重新聚合:通过蒸馏,将降解产物除去溶剂后,加入二正丁基镁(0.054毫升,0.054毫摩尔),-60℃反应24小时。反应结束后,加入苯甲酸/二氯甲烷溶液溶解混合物,取出加入到冷的甲醇溶液中,有聚合物析出。过滤分离得到白色固体,转移至真空干燥箱中干燥,得到聚合物。转化率通过反应液1hnmr计算得到,聚合物的分子量及分散度通过gpc测定。六氢-(3,4)-反式苯并呋喃酮转化率为80%,γ-丁内酯转化率为80%,聚合物数均分子量为9020克/摩尔,分散系数为1.19。
制备的功能化聚合物完全降解至单体,无需进一步提纯,降解获得的单体通过重新聚合,可制备功能化聚合物,实现了单体-聚合物-单体的化学循环。
- 还没有人留言评论。精彩留言会获得点赞!