一种环状碳酸酯的制备方法与流程
本发明属于绿色催化合成技术领域,具体涉及一种环状碳酸酯的制备方法。
背景技术:
目前,对二氧化碳(co2)的转化利用已成为全球普遍关注的焦点。已报道的关于co2的转化途径很多,但由于co2分子自身十分稳定,很难与其它物质发生反应,因此以co2为原料实现工业化生产的工艺路线很少。而由co2与环氧化物合成环状碳酸酯的路线正是其中之一。
以碳酸丙烯酯/碳酸乙烯酯为代表的环状碳酸酯是性能优良的高沸点极性有机溶剂,广泛应用于纺织、电池、化妆品、气体分离以及天然气和合成氨原料气中的二氧化碳(co2)和硫化氢气体的吸收等领域。它也是一种重要的有机化学品,用于制备聚碳酸酯及精细化工品。环状碳酸酯的合成方法主要包括光气法、酯交换法、尿素醇解法、co2与环氧化物环加成反应法等。其中co2与环氧化物环加成反应合成环状碳酸酯是典型的原子经济性反应,符合绿色化学的发展方向,其研究开发受到广泛重视。
目前报道的关于co2与环氧化物合成环状碳酸酯的催化剂很多,主要有季铵盐、碱金属卤化物、有机磷盐、离子液体、过渡金属配合物、金属氧化物、分子筛、负载型金属卤化物、负载型金属配合物等(a.a.g.shaikh,etal.,chem.rev.,96(1996),951;t.sakakura,etal.,chem.rev.,107(2007),2365;t.sakakura,etal.,chem.commun.,2009,1312;k.yamaguchi,etal.,j.am.chem.soc.,121(1999)4526;m.shi,j.-w.huang,j.org.chem.,68(2003),6705;v.calo,a.nacci,etal.,org.lett.,4(2002),2561;k.mori,etal.,chem.comm.,2005,3331;s.udayakumar,etal.,cata.commun.,10(2009),659;b.m.bhanage,etal.,greenchem.,5(2003),71.邓友全等,cn1343668;杨彩虹等,cn1424147;吕小兵等,cn1189246)。虽然现有的催化剂活性相对较高,但是仍需不断从绿色、便宜、高效等方面改进催化剂。于是开发一些高效、绿色、便宜的催化剂成为催化环状碳酸酯反应的一个方向。
技术实现要素:
为了解决上述问题,本发明提供一种可以在相对温和的条件下高速率、高收率的由二氧化碳和环氧化物合成环状碳酸酯的方法,该方法所用催化剂不含金属、催化效率更高。
为了解决上述技术问题,本发明的具体方案如下:
一种环状碳酸酯的制备方法,式iii或iv所示的环氧化物和二氧化碳在式i或式ii所示的催化剂催化下得到环状碳酸酯
r1、r2、r3选自氢、具有1~4个碳原子的直链或支链的烷基中相同或不同的取代基;
r4、r5选自氢、具有1~4个碳原子的直链或支链的烷基中相同或不同的取代基;
r6、r7选自氢、具有1~5个碳原子的直链或支链的烷基、具有1~5个碳原子并被卤素、羟基、苯酚基、乙烯基、烯丙氧基或具有1~4碳原子的烷氧基取代的直链或支链的烷基、苯基、卤代苯基;
n选自1~2的整数;
m选自o或-ch2-。
优选的,所述r1、r2、r3选自氢、具有1~4个碳原子的直链烷基中相同或不同的取代基;
r4、r5选自氢选自氢、甲基中相同或不同的取代基。
优选的,所述的式i或式ii所示的催化剂选自如下结构:
所述的式iii或iv所示的环氧化物选自如下结构:
优选的,所述制备方法的反应温度为20-140℃,二氧化碳的初始压力为0.1-5mpa,催化剂的用量是0.1-10mol%。
优选的,所述制备方法的反应温度为120-140℃,二氧化碳的初始压力为0.1-4mpa,催化剂的用量是2-5mol%。
优选的,所述二氧化碳的初始压力为0.2mpa。
优选的,所述的制备方法的具体步骤包括:
(1)式iii或iv所示的环氧化物和二氧化碳在式i或式ii所示的催化剂加入反应容器,二氧化碳置换反应容器中的空气;
(2)向反应容器中充入co2至初始压力为0.1-5mpa,升温至20-140℃;
(3)反应1~24h,冷却,得到环状碳酸酯。
本发明所述的发明方法的机理是:铵卤化物与环氧化物上的氧形成氢键,降低环氧化物上氧的电子云密度。对于季胺盐类催化剂来说,对环氧化物起活化作用的是两个羟基;对于叔胺盐类催化剂来说,对环氧化物起活化作用的是两个羟基和n-h键,分析实验结果,发现含n-h键的催化剂的反应条件相对来说比较温和,且反应速率较高。卤素负离子作为亲核试剂进攻碳-氧键,使环氧化物开环。铵卤化物活化开环的环氧化物末端氧负离子,氧负离子作为亲核试剂进攻二氧化碳,形成碳酸酯化合物。
有益效果
(1)本发明所用催化剂合成方法简单,更加易得。
(2)催化剂催化体系简单,催化活性更高,产物选择性好,催化反应条件温和安全,催化后所得产物无金属残留,且催化剂可以重复使用。
(3)铵卤化物催化反应,过程简单,所需要的设备简便,适用于工业化放大。
(4)反应过程不需要使用溶剂,避免有机溶剂的毒性,易于后期分离,达到绿色化学工艺。
(5)本发明所述的优选温度为20~140℃,更加优选温度为120-140℃,其主要原因在于:反应速度和温度成正比。温度越高,反应越快。但是当温度过高的时候,能耗过多,且反应选择性降低。而温度过低的时候,反应速度较慢且对催化剂的用量需求增多。经过实验得到综合反应时间,速度,选择性和能耗,最佳的反应温度在120-140℃。
(6)本发明所述的优选压力为0.1-5.0mpa,更加优选的压力为0.1-4.0mpa,其主要原因在于:二氧化碳的压力和反应速度成正比,压力越高,反应越快。但是当压力过高的时候,对于设备的耐压性能需求较高。在综合比较耐压设备、催化剂用量、温度等外界条件下,0.1mpa-4.0mpa是最适合的反应条件。
(7)本发明所述的最终优选压力为0.2mpa。
综上所述,本发明相比现有的催化体系具有简单、温和、高效、不含金属等明显的优势。
附图说明
图1:实施例5产物的氢谱图
图2:实施例9产物的氢谱图
图3:实施例4产物的氢谱图
图4:实施例17催化剂产物的氢谱图
具体实施方式
通过下列实施例可以进一步说明本发明,实施例是为了说明而非限制本发明的。本领域的任何普通技术人员都能够理解这些实施例不以任何方式限制本发明,可以对其做适当的修改和数据变换而不违背本发明的实质和偏离本发明的范围。
说明书中所涉及的各种原料,均购自市场,实施例中所用催化剂是根据现有技术合成所得,实施例中所用超导核磁共振波谱仪型号为brukerascendtm400,实施例中转化率由核磁测得。
实施例中所用的催化体系的结构如下:
实施例中所用的环氧化物的结构如下:
结构与编号
实施例1:
n-h催化:将环氧化物13(10mmol)、铵卤化物催化剂1(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.1mpa,后密封反应釜,升温至20℃,反应时间24h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率86%,选择性97%的淡黄色油状物质。
实施例2:
n-h催化:将环氧化物24(10mmol)、铵卤化物催化剂2(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至120℃,反应时间4h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率91%,选择性99%的淡黄色油状物质。
实施例3:
n-h催化:将环氧化物15(10mmol)、铵卤化物催化剂3(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至90℃,反应时间2h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率78%,选择性92%的淡黄色油状物质。
实施例4:
n-h催化:将环氧化物24(10mmol)、铵卤化物催化剂8(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至120℃,反应时间1.5h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率97%,选择性88%。1hnmr(400mhz,dmso-d6)1hnmrδ7.47(tt,j=8.1,4.7hz,5h),5.87(t,j=8.0hz,1h),4.89(t,j=8.3hz,1h),4.42(t,j=8.2hz,1h).产物的氢谱图如图3所示。
实施例5:
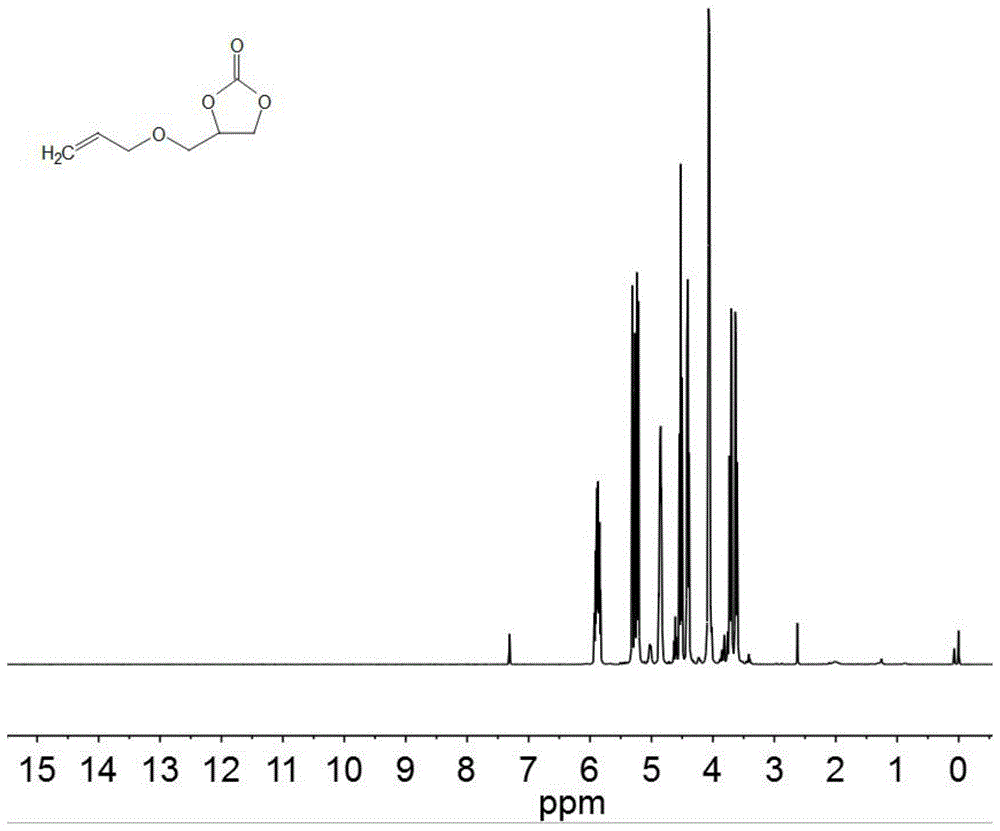
n-h催化:将环氧化物23(10mmol)、铵卤化物催化剂10(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至120℃,反应时间1h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率82%,选择性89%。产物的氢谱图如图1所示,(1hnmr(400mhz,cdcl3)δ5.94-5.81(m,1h),5.35-5.14(m,2h),4.86(dqd,j=9.1,4.8,4.2,2.1hz,1h),4.52(td,j=8.4,1.4hz,1h),4.41(ddd,j=8.1,6.1,1.4hz,1h),4.06(dt,j=5.7,1.8hz2h),3.71(ddd,j=11.1,3.7,1.3hz,1h),3.62(ddd,j=11.1,3.8,1.3hz,1h)。
实施例6:
n-h催化:将环氧化物26(10mmol)、铵卤化物催化剂11(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至120℃,反应时间1.5h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率93%,选择性98%的白色固体。
实施例7:
羟基催化:将环氧化物17(10mmol)、铵卤化物催化剂6(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为5.0mpa,后密封反应釜,升温至50℃,反应时间4h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率86%,选择性98%的淡黄色油状物质。
实施例8:
羟基催化:将环氧化物18(10mmol)、铵卤化物催化剂7(0.5mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.1mpa,后密封反应釜,升温至140℃,反应时间3h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率90%,选择性98%的淡黄色油状物质。
实施例9:
羟基催化:将环氧化物22(10mmol)、铵卤化物催化剂9(0.01mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至90℃,反应时间4h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率71%,选择性98%。产物的氢谱图如图2所示,(1hnmr(400mhz,cdcl3),δ4.74-4.66(m,1h),4.40(td,j=8.2,1.2hz,1h),4.31(ddd,j=7.8,5.9,1.1hz,1h),3.54(ddd,j=10.4,4.5,1.1hz,1h),3.49-3.41(m,1h),1.12(d,j=1.3hz,9h)。
实施例10:
催化剂1的合成:在40℃下,将7.4ml的环氧丙醇滴加到含7.5ml的二乙胺反应瓶中,在氩气的保护下,反应5h,之后用旋蒸除去多于的二乙胺。再向处理过的反应中滴加等量的氢碘酸,之后用50ml的乙醚和丙酮分别洗涤5次,得到淡黄色油状物质,真空干燥24h,即可得到催化剂1。
实施例11:
催化剂2的合成:在60℃下,将7.4ml的环氧丙醇滴加到含11ml的二丙胺的反应瓶中,在氩气的保护下,反应5h,之后用旋蒸除去多于的二丙胺。再向处理过的反应中滴加等量的氢碘酸,之后用50ml的乙醚和丙酮分别洗涤5次,得到淡黄色油状物质,真空干燥24h,即可得到催化剂2。
实施例12:
压力对反应的影响:在不同的压力条件下探究压力对实验结果的影响。具体的实施方法如下:参照实施例4的实验步骤,在120℃,2mol%催化剂8负载量,1h反应时间的条件下,分别采用不同的反应压力:0.1mpa,0.2mpa,0.4mpa,0.5mpa,0.6mpa,0.8mpa,1.0mpa,1.5mpa,2mpa来实施实验。并获得不同压力条件下实验最终的环碳酸酯的转化率如下表所示)。通过分析实验数据,我们发现当反应压力由1个大气压增加到2个大气压时,反应速率相应提高了2.3倍。再继续增加反应压力直至20个大气压,我们发现反应速率几乎没有改变。
实施例13:
羟基催化:将环氧化物13(10mmol)、铵卤化物催化剂6(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.1mpa,升温至20℃,反应时间24h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率62%,选择性87%的淡黄色油状物质。
由实施例1、2、3、4、5、6和实施例7、8、9的实验数据可看出,含n-h键的催化剂的催化活性更高;由实施例1、2、3、4、5、6的实验数据看出,催化剂8的催化活性最好;由实施例1、2、3和实施例4、5、6的对比可粗略看出,催化剂的r取代基为环状时的催化效果更好。由实施例12的实验数据看出,实验的压力对反应的转化率有较大的影响,反应的最佳压力为0.2mpa,若再增加反应压力,反应的转化率没有较大的提高。
实施例14
n-h催化:将环氧化物24(10mmol)、铵卤化物催化剂8(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为4.0mpa,后密封反应釜,升温至60℃,反应时间6h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率86%,选择性90%。
实施例15
n-h催化:将环氧化物24(10mmol)、铵卤化物催化剂8(1.0mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.1mpa,后密封反应釜,升温至120℃,反应时间2h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率97%,选择性96%。
实施例16
n-h催化:将环氧化物28(10mmol)、铵卤化物催化剂8(0.2mmol)加入带有机械搅拌和温控加热装置的高压釜中。密封反应釜,用co2置换釜中空气3次,然后向反应釜中充入co2至初始压力为0.2mpa,后密封反应釜,升温至60℃,反应时间2h。待反应完成后,用冰水混合物冷却反应釜至0℃,释放出残余气体,采用气相色谱进行定性定量分析,得到转化率79%,选择性96%,生成的环碳酸酯产物为淡黄色油状物质。
实施例17
催化剂8的合成:在25℃下,将7.4ml的环氧丙醇滴加到含7.5ml的吡咯烷的反应瓶中,在氩气的保护下,反应1h,之后用旋蒸除去多于的吡咯胺。再向处理过的反应中滴加等量的氢碘酸,之后用50ml的乙醚和丙酮分别洗涤5次,得到淡黄色油状物质,真空干燥24h,即可得到催化剂8。产物的氢谱图如附图4所示,1hnmr(400mhz,dmso-d6)δ5.55(s,1h),4.94(t,j=5.5hz,1h),3.79(ddd,j=9.2,5.5,2.9hz,1h),3.57–3.04(m,6h),1.93(d,j=39.4hz,4h),1.23(s,1h)。
- 还没有人留言评论。精彩留言会获得点赞!