一种双核稀土烯烃聚合催化剂及其应用的制作方法
本发明技术领域涉及一种双核稀土烯烃聚合用催化剂,尤其涉及一种路易斯碱稳定的双核双茂稀土烯烃聚合用催化剂及其应用。
背景技术:
烯烃类橡胶用催化剂体系是烯烃橡胶生产技术的关键,目前采用的催化剂体系主要有镍(ni)系、钛(ti)系、钴(co)系、稀土钕(nd)系以及锂(li)系等(姜连升等,稀土顺丁橡胶.冶金工业出版社,2016,39)。其中,稀土催化剂又是最具特色且综合性能优良的品种,用其生产的烯烃橡胶顺式结构含量高、线形结构规整度高、分子量高且分布窄。
1970年中国科学院长春应化所首先研制成功了制备高顺式聚丁二烯橡胶用的稀土羧酸盐、烷基铝和氯化烷基铝的三元催化体系(稀土催化合成橡胶文集[c].科学出版社,1980,25)。20世纪80年代,德国bayer公司(at133rdmeetingoftherubberdivisionofacs,1988,4,19;at133rdmeetingoftherubberdivisionofacs,1988,10,18)和意大利enichem公司(kautschukgummikunststiffe,1993,6,458)先后实现了稀土顺丁橡胶的工业化。目前工业上使用的稀土催化剂仍主要为三元钕系催化剂,其核心技术主要掌握于德国lanxess公司(ep2311889、ep2363303、ep2676968、ep3057998、cn102574955、cn102762613、cn104395351、cn107254008)以及长春应化所张学全研究组(cn01128284、cn01128287、us7288611、cn01128289、cn03127180、cn200610016949)。
目前,用于共轭二烯烃定向聚合的具有特定结构的单活性中心稀土金属催化剂的研究得到了较好的发展,其催化二烯烃聚合反应具有高活性、窄分子量分布的特征(j.organomet.chem.,2001,621,327;macromol.chem.phys.,2003,204,1747;macromol.chem.phys.,2004,205,737;angew.chem.,2005,117,2649;angew.chem.int.ed.,2005,44,2593;macromolecules1999,32,9078;macromolecules2001,34,1539;macromolecules2003,36,7923;macromolecules2004,37,5860;macromolecules2006,39,1359-1363;daltontrans.,2008,2531;angew.chem.int.ed.2007,46,1909;j.am.chem.soc.2008,130,4984.)。
由于稀土金属元素通常为+3价,因此用于烯烃聚合的阳离子型单活性中心稀土催化剂通常为单茂或非茂型结构,双茂稀土金属催化剂由于阳离子化后没有插入反应位点,理论上来说没有催化活性。我们通过双路易斯碱对2分子的单核双茂稀土络合物进行桥接,希望通过双金属间的协同作用或改变金属-配体间的配位方式来达到提高催化活性的目的。之前报道的一些双核或多核稀土金属络合物通常通过h或烷基的σ键配位来进行桥接(macromolecules2004,37,5860;macromolecules2006,39,1359-1363;chemcomm,2015,51,5063-5065),或通过苯基、二茂铁等基团对两个一价配体进行连接来实现(cn201210117207,cn201711172220),而通过加入双官能化路易斯碱将双茂稀土金属络合物进行桥接尚未见报道。
技术实现要素:
本发明的目的在于提供一种双核稀土烯烃聚合催化剂,具体为路易斯碱稳定的双核双茂稀土催化剂组合物,该催化剂组合物特别适用于高顺式共轭二烯类橡胶的制备。
为达上述目的,本发明提供的双核稀土烯烃聚合催化剂,由主催化剂、助催化剂、链转移试剂组成;主催化剂为路易斯碱稳定的双核稀土络合物,由化学式[i]和化学式[ii]表示:
其中,ln为第iiib族的过渡金属元素;
x选自三甲基甲硅烷基甲基、二(三甲基甲硅烷基)甲基、三(三甲基甲硅烷基)甲基、邻-(n,n-二甲氨基)苄基、n,n-二(三甲基甲硅烷基)胺基中的一种;
r1至r18相同或不同,各自独立地选自氢、c1-c10的饱和烷基中的至少一种;
助催化剂选自化学式[iii]所示化合物;化学式[iii]表示为:[eh]+[ba4]-、[e]+[ba4]-或ba3,其中,e是含氮或碳的中性或阳离子型路易斯酸,b是硼元素,h为氢元素,a为选自c6-c30芳基或卤代芳基、c1-c10烷基或卤代烷基中的至少一种。
链转移试剂选自化学式[iv]所示化合物;化学式[iv]表示为:al(r19)(r20)(r21),其中,al为铝元素,r19至r21相同或不同,各自独立地选自氢、卤素、c1-c10的烷基或卤代烷基中的至少一种。
本发明的双核稀土烯烃聚合催化剂,其中,ln选自钪、钇以及镧系稀土金属元素中的至少一种,进一步优选为y和gd。
本发明的双核稀土烯烃聚合催化剂,其中,
本发明的双核稀土烯烃聚合催化剂,其中,x选自三甲基甲硅烷基甲基、二(三甲基甲硅烷基)甲基、三(三甲基甲硅烷基)甲基、邻-(n,n-二甲氨基)苄基、n,n-二(三甲基甲硅烷基)胺基中的一种,但不限于此;进一步优选为邻-(n,n-二甲氨基)苄基。
本发明的双核稀土烯烃聚合催化剂,其中,r1至r18相同或不同,各自独立地选自氢、c1-c10的饱和烷基;其中,c1-c10的饱和烷基优选自甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基、戊基、己基、庚基和辛基中的至少一种,但不限于此;进一步优选为甲基。
本发明的双核稀土烯烃聚合催化剂,其中,化学式[i]和化学式[ii]表示的双核稀土络合物为原位生成,通过单核稀土络合物与双路易斯碱化合物之间的配位反应合成,并直接用于聚合反应。
本发明的双核稀土烯烃聚合催化剂,其中,双核稀土络合物生成时,双路易斯碱与单核稀土络合物的摩尔比为0.5/1。
本发明的双核稀土烯烃聚合催化剂,其中,化学式[iii]所示化合物优选自三苯基(甲基)-四(五氟苯)硼盐、苯基-二甲基氨基-四(五氟苯)硼盐、苯基-二甲基氨基-四苯基硼盐、三(五氟苯)硼盐、三苯基硼盐中的至少一种,进一步优选为苯基-二甲基氨基-四苯基硼盐。
本发明的双核稀土烯烃聚合催化剂,其中,化学式[iv]所示化合物优选自三甲基铝、三乙基铝、三正丙基铝、三正丁基铝、三异丙基铝、三异丁基铝、三己基铝、三环己基铝、三辛基铝、三苯基铝、三对甲苯基铝、三苄基铝、乙基二苄基铝、二乙基苄基铝、乙基对甲苯基铝中的至少一种,但不限于此;进一步优选为三异丁基铝。
本发明的双核稀土络合物可以按照路线c进行制备:
(a)
(b)
如图所示,路线(a)为非桥联双核稀土络合物的合成,路线(b)为硅桥联双核稀土络合物的合成。通过单核的稀土络合物与0.5当量的双路易斯碱反应,反应溶剂为甲苯,反应温度为25℃,反应完成后不进行分离,直接与助催化剂作用后用于烯烃聚合反应。
本发明的双核稀土烯烃聚合催化剂,其中,化学式[ⅲ]所示化合物与主催化剂的摩尔比优选为0.1/1至10/1,进一步优选为1.0/1至2.0/1;化学式[ⅳ]所示化合物与主催化剂的摩尔比优选为1/1至10000/1,进一步优选为10/1至1000/1。
本发明还提供一种上述双核稀土烯烃聚合催化剂在制备高顺式共轭二烯类橡胶中的应用。
本发明的双核稀土烯烃聚合催化剂中,化学式[iv]表示的烷基铝试剂可以提前加入反应体系中与聚合单体混合。化学式[iv]表示的化合物主要是作为聚合物链转移试剂使用,其用量对聚合物的分子量有较大影响。此外,其还作为主要的除杂试剂与聚合单体中的杂质进行反应除杂。因此,当化学式[iv]表示的化合物/助催化剂的摩尔比在上述范围时,可以避免由于聚合单体无法彻底除杂而导致的催化剂部分失活的问题;此外,还可以控制聚合物分子量分布在合适的范围内。
本发明的双核稀土烯烃聚合催化剂用于制备共轭二烯聚合物,该制备过程为均相的溶液聚合,聚合溶剂通常为正己烷,本发明提供的双核稀土烯烃聚合催化剂可以高活性地催化1,3-丁二烯的聚合,聚合反应制得的聚丁二烯橡胶具有较高的顺式1,4-含量以及较窄的分子量分布;且与对应的单核稀土催化剂相比,本发明的双核稀土烯烃聚合催化剂具有更高的催化活性,且聚合反应生成的烯烃聚合物具有更高的立构规整度。
附图说明
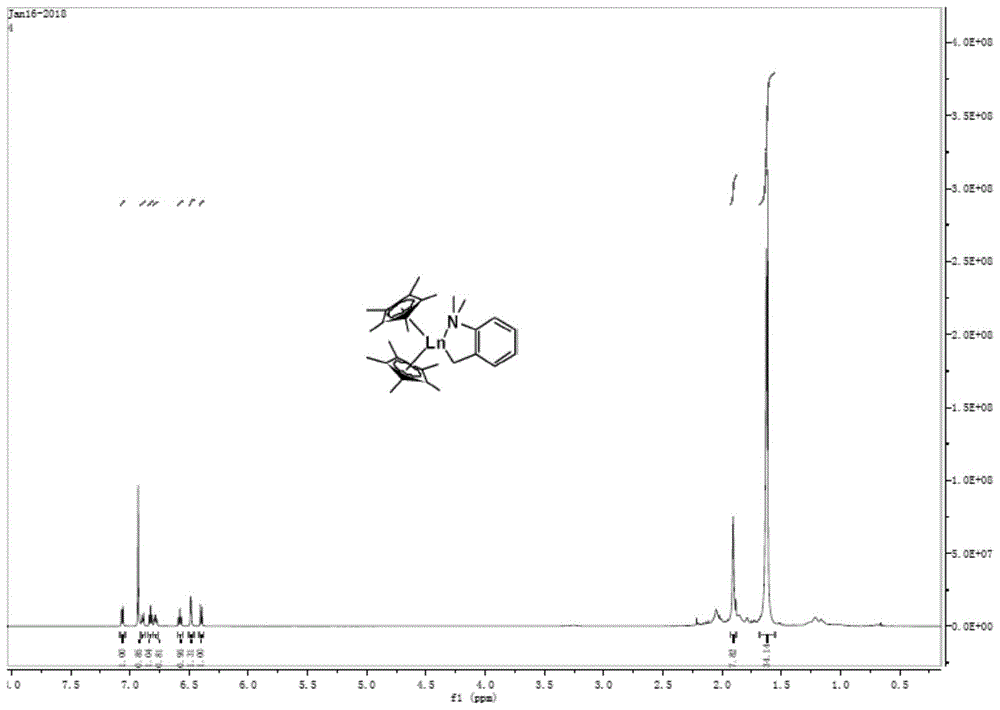
图1为实施例1所示化合物的1h-nmr谱图;
图2为实施例2所示化合物的1h-nmr谱图;
图3为实施例5所示化合物的1h-nmr谱图。
具体实施方式
以下列举较佳实施例对本发明作详细说明,虽然以下实施例在以本发明提供的技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围并不限于下述实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
其中,部分单核稀土络合物的合成可参考文献(journaloforganometallicchemistry,322(1987)321-329;macromolecules2004,37,5860-5862;organometallics,2005,24,2269-2278)进行合成。
以下实施例中,双核稀土烯烃聚合催化剂的合成以及催化烯烃聚合反应如无特殊说明均在无水无氧条件下进行,通过惰性气体手套箱或schlenk技术实现,实验所用溶剂均经过无水无氧处理;但本发明并不限于此。
此外,稀土金属配合物的核磁共振1h-nmr谱图通过brukerascend600mhz进行测试,部分化合物由于顺磁特性无法进行1h-nmr表征。聚合物的顺式-1,4选择性通过13c-nmr谱图确定,通过反门控去耦合方式进行测定;聚合物的分子量及分子量分布通过pl-gpc50凝胶渗透色谱仪进行测试。
实施例1
单核稀土络合物1的制备
0℃下,将正丁基锂缓慢滴加到cp*的己烷溶液中,随后自然升至室温并搅拌过夜,产生大量淡黄色沉淀。随后,于惰性气体手套箱中进行抽滤,所得固体用己烷洗涤2次,真空下干燥后得到淡黄色粉末固体即为cp*li化合物。室温下,将cp*li的thf溶液缓慢滴加到ycl3.nthf的thf悬浊液中,随后加热回流过夜。反应结束后,抽干thf溶液,加入甲苯萃取。将甲苯萃取液浓缩后置于-30℃下重结晶,得到白色固体即为稀土氯化物cp*2ycl2li(thf)2。
-30℃下,将邻二甲胺基苄基锂(abzli)的甲苯溶液缓慢滴加到cp*2ycl2li(thf)2的甲苯溶液中,自然升至室温后继续反应4h。反应完成后用砂芯漏斗进行过滤,滤液在真空下抽干后得到黄色油状物,用少量正己烷洗涤后得到淡黄色粉末,即为cp*2yabz产物(化合物1)。
核磁数据:1h-nmr(600mhz,c6d6):1.86(s,30h,cp-me),2.14(s,6h,n-me),2.28(br,2h,y-ch2),6.71(d,j=6hz,1h),6.81(t,j=6hz,2h),7.05(t,j=6hz,2h),7.29(d,j=6hz,1h)。
实施例2
单核稀土络合物2的制备
-30℃下,将烷基锂(tmsch2li)的甲苯溶液缓慢滴加到cp*2ycl2li(thf)2的甲苯溶液中,自然升至室温后继续反应4h。反应完成后用砂芯漏斗进行过滤,滤液在真空下抽干后得到黄色油状物,用少量正己烷洗涤后得到淡黄色粉末,即为cp*2yach2tms产物(化合物2)。
核磁数据:1h-nmr(600mhz,c6d6):0.00(s,2h,ch2tms),0.44(s,9h,sime3),1.98(s,30h,cp-me)。
实施例3
单核稀土络合物3的制备
化合物3的合成参照化合物1的合成,将起始原料中的ycl3.nthf换成gdcl3.nthf,实验步骤一致,所得产物为淡黄色粉末固体。由于+3价gd化合物顺磁性的特征,无法进行nmr表征,仅对cp*2gdcl2li(thf)2进行了x-射线单晶衍射表征。
实施例4
单核稀土络合物4的制备
化合物4的合成参照化合物2的合成,将起始原料中的ycl3.nthf换成gdcl3.nthf,实验步骤一致,所得产物为淡黄色粉末固体。由于+3价gd化合物顺磁性的特征,无法进行nmr表征,仅对cp*2gdcl2li(thf)2进行了x-射线单晶衍射表征。
实施例5
单核稀土络合物5的制备
-30℃下,将正丁基锂(2.4minn-hexane)缓慢滴加到二甲硅基me2si桥联的双(四甲基环戊二烯)配体的乙醚溶液中,随后升至室温并继续搅拌2小时,反应结束后真空下抽干溶剂得到淡黄色固体即为配体的双锂盐。随后在-30℃下,将双锂盐的thf溶液缓慢滴加到ycl3.(thf)n的thf悬浊液中,滴加完成后将反应溶液加热至回流,并继续反应过夜。反应结束后,真空下抽干thf,加入甲苯萃取,过滤后将滤液抽干并用少量无水己烷洗涤,得到白色固体粉末即为si(cpme4)2ycl2li(thf)2。
-30℃下,将苄基锂的甲苯悬浊液缓慢加入茂稀土金属卤化物的甲苯悬浊液中,升至室温后继续反应过夜,反应完成后过滤除掉生成的licl,滤液在真空下取出溶剂后,用少量无水正己烷洗涤两遍得到稀土苄基化合物si(cpme4)2yabz(化合物5)。
核磁数据:1h-nmr(600mhz,c6d6):0.94(s,3h,si-me),1.03(s,3h,si-me),1.39(s,2h,y-ch2),1.45(s,6h,cp-me),1.76(s,6h,cp-me),2.00(s,6h,n-me),2.07(d,12h,j=6hz,cp-me),6.56(m,1h,ar-h),6.75(m,1h,ar-h),7.02(m,1h,ar-h),7.19(m,1h,ar-h)。
实施例6
单核稀土络合物6的制备
-30℃下,将正丁基锂(2.4minn-hexane)缓慢滴加到二甲硅基me2si桥联的双(四甲基环戊二烯)配体的乙醚溶液中,随后升至室温并继续搅拌2小时,反应结束后真空下抽干溶剂得到淡黄色固体即为配体的双锂盐。随后在-30℃下,将双锂盐的thf溶液缓慢滴加到ycl3.(thf)n的thf悬浊液中,滴加完成后将反应溶液加热至回流,并继续反应过夜。反应结束后,真空下抽干thf,加入甲苯萃取,过滤后将滤液抽干并用少量无水己烷洗涤,得到白色固体粉末即为si(cpme4)2ycl2li(thf)2。
-30℃下,将lich2tms的甲苯溶液缓慢加入si(cpme4)2ycl2li(thf)2的甲苯悬浊液中,升至室温后继续反应过夜,反应完成后过滤除掉生成的licl,滤液在真空下取出溶剂后,用少量无水正己烷洗涤两遍得到稀土苄基化合物si(cpme4)2ych2tms(化合物6)。由于+3价gd化合物顺磁性的特征,无法进行nmr表征。
实施例7-10
室温下,将4,4’-联吡啶的甲苯溶液导入单核稀土络合物1-4的甲苯溶液中,溶液由淡黄色立即变成深黄色,随后室温下继续搅拌15min得到深棕色溶液,即为原位制备的双核稀土金属络合物7-10(化合物7-10),直接用于聚合实验。
实施例11-12
实施例11-12对应的双核稀土络合物的合成方法参照化合物7-10的合成,原位制备后直接用于聚合实验。
实施例13
催化剂聚合评价:
于惰性气体手套箱中,往100ml圆底烧瓶中称取1,3-丁二烯的己烷溶液(质量分数15%)14.4g(2.16g,40mmol),加入三异丁基铝的己烷溶液0.2ml(1m,0.2mmol),并于室温下搅拌30min,向聚合溶液中加入实施例7的双核稀土络合物(0.02mmol)的甲苯溶液及[phnhme2][b(c6f5)4]的甲苯悬浊液。室温下聚合30min后,聚合体系变粘稠,聚合单体完全消耗。聚合完成后,将反应瓶移出惰性气体手套箱,搅拌下缓慢加入无水甲醇直至聚合物完全析出,加入bht抗氧剂0.02g(聚合物质量的1%),用无水甲醇洗涤聚合物3次,置于真空烘箱中70℃烘干5小时,称重。
聚合结果见表1所示。
实施例14-16
催化剂聚合评价:
根据与实施例13中相同的方法进行丁二烯聚合,不同之处在于依次使用实施例8-10的双核稀土络合物。
聚合结果见表1所示。
实施例17-18
催化剂聚合评价:
根据与实施例13中相同的方法进行丁二烯聚合,使用实施例6的双核稀土络合物,不同之处在于丁二烯与稀土的摩尔比分别为10000和20000。
聚合结果见表1所示。
表1实施例13-18
说明:(1)[b]n=[phnhme2][b(c6f5)4],[b]n/[ln]=1.2
从表中数据可以看出,中心金属为y时催化活性较高,但顺式-1,4选择性低于90%;而中心金属为gd时,催化活性较低,但是顺式-1,4选择性可以达到99%。
比较例1-4
根据与实施例13中相同的方法进行丁二烯聚合,不同之处在于依次使用实施例1-4的单核稀土络合物。
聚合结果见表2所示。
表2比较例1-4
说明:(1)[b]n=[phnhme2][b(c6f5)4],[b]n/[ln]=1.2
通过对比(实施例13对比比较例1,实施例15对比比较例3)可以看出,使用单核稀土金属络合物作为助催化剂,在同等的聚合实验条件下,无论催化剂活性还是聚合产物的立体规整度都要远远低于双核稀土金属络合物。
如上所述,本发明提供的包含路易斯碱连接的双核双茂稀土金属络合物的双核稀土烯烃聚合催化剂在烯烃聚合反应中,对比相应的单核稀土络合物具有高活性和高立体选择性等优势。
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明所附的权利要求书的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于烯烃聚合的催化剂组分及其制备方法和应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂体系及其应用和烯烃聚合方法与制造工艺
- 卤化镁醇合物载体及其制备方法和应用以及用于烯烃聚合的催化剂组分及其应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺