mPEG-b-PLC两亲性嵌段共聚物的制备方法和多西紫杉醇纳米制剂及制备方法与流程
本发明属于生物医药
技术领域:
,涉及一种mpeg-b-plc两亲性嵌段共聚物的制备方法和多西紫杉醇纳米制剂及制备方法。
背景技术:
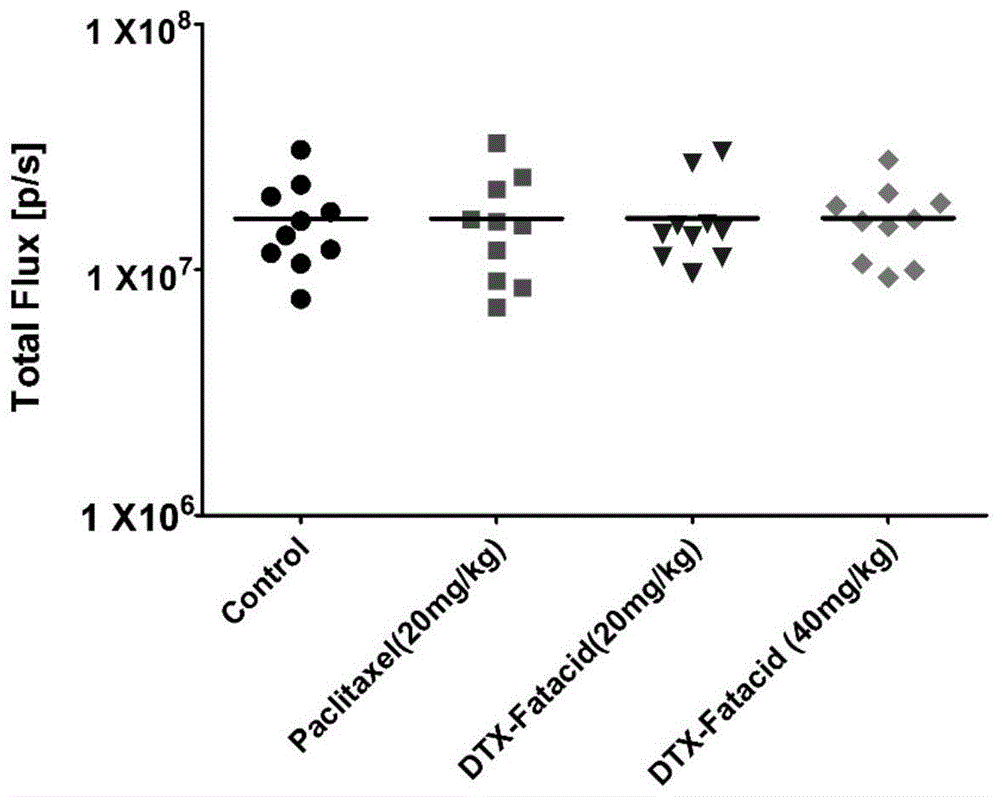
:多西紫杉醇是紫杉醇的衍生物,和紫杉醇一样具有抗肿瘤活性,其作用靶点在包浆的微管,抗瘤谱广。多西紫杉醇水溶性差,因此需要开发多西紫杉醇水溶性注射用剂,以便于给药。taxotere(泰索帝)是sanofi-aventis公司开发的多西紫杉醇注射液抗癌药,每毫升taxotere含有40mg多西紫杉醇、1040mg聚山梨酯80和13%乙醇,taxotere通常用于治疗乳腺癌、卵巢癌等。然而,taxotere易引发骨髓抑制、过敏、胃肠道反应、体液潴留、关节痛等毒副作用。这些毒副作用与药物助剂有关,因此改变助剂将有希望降低制剂的毒副作用。mpeg-b-plc两亲性嵌段共聚物可以作为药物助剂,但是现有制备技术制得的mpeg-b-plc两亲性嵌段共聚物具有分子量分布宽(分子量分布约为1.7左右)、锡元素含量较高(400~600ppm)的特点,锡元素属于重金属且亚锡离子易被氧化,如不对其处理一旦进入体内会危害人体的健康。技术实现要素:本发明的目的是针对现有mpeg-b-plc两亲性嵌段共聚物和多西紫杉醇制剂问题,提供新型的mpeg-b-plc两亲性嵌段共聚物制备方法、提供毒副作用低的新型多西紫杉醇纳米制剂及其制备方法。为了实现上述目的,本发明提供如下技术方案:一种mpeg-b-plc两亲性嵌段共聚物的制备方法,包括如下步骤:(1)真空干燥mpeg;(2)混合mpeg与异辛酸亚锡,加入dl-丙交酯,在n2氛围中于160~180℃条件下反应3~5小时,获得反应物;(3)冷却至室温,在n2氛围下,将反应物加入乙酸乙酯和正庚烷的混合溶剂中,完全溶解后,加入无水乙醚以去除未反应的丙交酯及反应生成的丙交酯均聚物,过滤获得mpeg-b-plc两亲性嵌段共聚物粗品;(4)真空干燥mpeg-b-plc两亲性嵌段共聚物粗品;(5)将mpeg-b-plc两亲性嵌段共聚物粗品溶于水中,加入edta二钠与其中锡离子络合形成络合物,用透析袋透析20~50小时去除络合物,干燥获得mpeg-b-plc两亲性嵌段共聚物;或者将mpeg-b-plc两亲性嵌段共聚物粗品溶于水中,加入透析袋中,在透析介质中加入edta二钠与其中锡离子络合形成络合物,透析20~50小时去除络合物,干燥获得mpeg-b-plc两亲性嵌段共聚物;其中,所述mpeg-b-plc两亲性嵌段共聚物的锡元素含量为5~40ppm。优选地,所述步骤(1)具体为:将mpeg在50~60℃条件下真空干燥3~5小时;所述步骤(2)具体为:将异辛酸亚锡配制成异辛酸亚锡的甲苯溶液或异辛酸亚锡的二氯甲烷溶液,与mpeg混合后,密封,在70℃条件下吹入高纯n2以形成n2氛围,再加入dl-丙交酯,在n2氛围中于160~180℃条件下反应3~5小时,获得反应物;所述步骤(2)中异辛酸亚锡的甲苯溶液或异辛酸亚锡的二氯甲烷溶液加入量为使异辛酸亚锡的质量为mpeg的0.2~1%;所述步骤(2)中mpeg与dl-丙交酯的质量比为1:(0.5~1)。优选地,所述步骤(3)中反应物与乙酸乙酯和正庚烷混合溶剂的体积比为1:(1~2);所述步骤(4)中真空干燥温度为35~45℃,时间为25~35小时。优选地,所述步骤(5)中,edta二钠以edta二钠水溶液的方式加入,edta二钠水溶液的浓度为0.0075g/l~0.15g/l,加入量为使edta二钠的质量为mpeg-b-plc两亲性嵌段共聚物粗品质量的1~2%;所述“用透析袋进行透析20~50小时”具体为:将待透析的溶液装入透析袋中,放入透析介质透析20~50小时,每5~7小时更换一次透析介质;所述透析介质为超纯水。一种多西紫杉醇纳米制剂,包括多西紫杉醇和权利要求1-4任一所述制备方法制得的mpeg-b-plc两亲性嵌段共聚物;所述多西紫杉醇与所述mpeg-b-plc两亲性嵌段共聚物的质量比为1:3~100。在本发明的具体实施方案中,多西紫杉醇与mpeg-b-plc两亲性嵌段共聚物的质量比可以为例如1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10、1:15、1:20、1:25、1:30、1:35、1:40、1:45、1:50、1:55、1:60、1:65、1:70、1:75、1:80、1:85、1:90、1:95、1:96、1:97、1:98、1:99、1:100等。一种上述多西紫杉醇纳米制剂的制备方法,包括如下步骤:s1、将多西紫杉醇溶解于第一有机溶剂,得到第一溶液;s2、将mpeg-b-plc两亲性嵌段共聚物溶解于第二有机溶剂,得到第二溶液;s3、将脂肪酸溶解于第三有机溶剂,得到第三溶液;s4、混合第一溶液、第二溶液和第三溶液,去除有机溶剂,获取混合物;s5、水化所述混合物,用纳米过滤器过滤,获取滤液;s6、冷冻所述滤液,获取多西紫杉醇纳米冰块;s7、冷冻干燥所述多西紫杉醇纳米冰块,获取多西紫杉醇纳米制剂;其中,所述多西紫杉醇和所述mpeg-b-plc两亲性嵌段共聚物的质量比为:1:3~100。优选地,所述第一有机溶剂、第三有机溶剂为无水乙醇;所述第二有机溶剂为乙腈;所述脂肪酸选自月桂酸、棕榈酸、油酸、亚油酸中任一种。优选地,步骤s4具体为:将所述第一溶液、第二溶液和第三溶液于60℃水浴下搅拌混合30分钟,再在60℃温度下负压去除有机溶剂,获得紫杉醇和mpeg-b-plc两亲性嵌段共聚物的混合物。优选地,步骤s5具体为:用蒸馏水水化混合物,震动混合15~30分钟获得混合液,用200纳米过滤器过滤混合液,获得滤液。优选地,步骤s6中冷冻所述滤液的温度为-40℃;步骤s7具体为:将紫杉醇纳米冰块用冻干机干燥48~72小时,获取多西紫杉醇纳米制剂。本发明技术方案制备的mpeg-b-plc两亲性嵌段共聚物具有锡元素含量低的优点,本发明多西紫杉醇纳米制剂具有毒副作用低、抗肿瘤效果优越的效果以及制备工艺简单、生产成本低的优点。附图说明图1、本发明实验例,各组动物模型day1生物发光强度。图2、本发明实验例,各组动物模型day1生物发光2d成像。图3、本发明实验例,各组动物模型day8生物发光强度。图4、本发明实验例,各组动物模型day8生物发光2d成像。图5、本发明实验例,各组动物模型day15生物发光强度。图6、本发明实验例,各组动物模型day15生物发光2d成像。图7、本发明实验例,各组动物模型day22生物发光强度。图8、本发明实验例,各组动物模型day22生物发光2d成像。图9、本发明实验例,各组动物模型生物发光强度增长倍数对比。图10、本发明实验例,各组动物模型体重增长倍数的比较。具体实施方式为使技术人员更好地理解本发明,下面对本发明的实施例进行清楚、详细的说明,但不作为对本发明的限定。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例1mpeg-b-plc两亲性嵌段共聚物的制备本实施例mpeg-b-plc两亲性嵌段共聚物的制备步骤如下:(1)将10gmpeg(分子量为5000)在温度60℃下真空干燥3小时;(2)将mpeg与0.1ml异辛酸亚锡的二氯甲烷溶液(异辛酸亚锡的质量体积百分含量为20%,即100ml溶液中含异辛酸亚锡20g)混合后,密封,然后在70℃下吹高纯n22小时,以形成n2氛围,加入10gdl-丙交酯,混合均匀后再于温度180℃条件下反应3小时得反应物;(3)在n2气氛下冷却至室温,将反应物加入到2倍体积的乙酸乙酯和正庚烷混合溶剂中,完全溶解后,按体积计加入10倍的无水乙醚使未反应的丙交酯及反应生成的丙交酯均聚物溶解,然后过滤得mpeg-b-plc两亲性嵌段共聚物粗品;(4)将mpeg-b-plc两亲性嵌段共聚物粗品在40℃温度条件下真空干燥30小时;(5)将干燥后的mpeg-b-plc两亲性嵌段共聚物粗品溶于水中,并加入浓度为0.0075g/l的edta二钠溶液,加入量为使edta二钠的质量占mpeg-b-plc两亲性嵌段共聚物粗品质量的1%,使亚锡离子络合形成络合物,然后装入到透析袋中,放入超纯水中透析30小时,每6小时更换一次超纯水,透析后冷冻干燥30小时得到本实施例mpeg-b-plc两亲性嵌段共聚物。实施例2mpeg-b-plc两亲性嵌段共聚物的制备本实施例中,mpeg-b-plc两亲性嵌段共聚物的制备步骤如下:(a)将10gmpeg(分子量为2000)在温度35℃下真空干燥8小时;(b)将mpeg与0.1ml异辛酸亚锡的甲苯溶液混合后,密封,在70℃下吹高纯n22小时,以形成n2氛围,加入5gdl-丙交酯,混合均匀后再于160℃条件下反应5小时得反应物;(c)在n2气氛下,冷却至室温,然后将反应物加入到与2倍体积的乙酸乙酯和正庚烷(2:1)混合溶剂中,完全溶解后,按体积计加入10倍的无水乙醚使未反应的丙交酯及反应生成的丙交酯均聚物溶解,然后过滤得mpeg-b-plc两亲性嵌段共聚物粗品;(d)将mpeg-b-plc两亲性嵌段共聚物粗品在35℃温度条件下真空干燥35小时;(e)将干燥后的mpeg-b-plc两亲性嵌段共聚物粗品溶于水中,并加入浓度为0.15g/l的edta二钠溶液,加入量为使edta二钠的质量占mpeg-b-plc两亲性嵌段共聚物粗品质量的2%,使亚锡离子络合形成络合物,然后装入到透析袋中,放入超纯水中透析20小时,每5小时更换一次超纯水,透析后冷冻干燥24小时得到本实施例mpeg-b-plc两亲性嵌段共聚物。实施例3mpeg-b-plc两亲性嵌段共聚物的制备本实施例中,mpeg-b-plc两亲性嵌段共聚物的制备步骤如下:1)将10gmpeg(分子量为2000)在温度45℃下真空干燥5小时;2)将mpeg与0.1ml异辛酸亚锡的甲苯溶液混合后,密封,然后在70℃下吹高纯n22小时,以形成n2氛围,加入7.5gdl-丙交酯,混合均匀后在于温度170℃条件下反应4小时得反应物;3)冷却至室温,将反应物加入到与2倍体积的乙酸乙酯和正庚烷(2:1)混合溶剂中,完全溶解后,加入10倍体积的无水乙醚使未反应的丙交酯及反应生成的丙交酯均聚物溶解,然后过滤得mpeg-b-plc两亲性嵌段共聚物粗品;4)将mpeg-b-plc两亲性嵌段共聚物粗品在45℃条件下真空干燥25小时;5)将干燥后的mpeg-b-plc两亲性嵌段共聚物粗品溶于水中,然后装入到透析袋中,透析介质采用edta二钠溶液,透析介质中edta二钠溶液的量为使edta二钠的质量占mpeg-b-plc两亲性嵌段共聚物粗品质量的1.5%,透析50小时,每7小时更换一次透析介质,透析后冷冻干燥40小时得到本实施例mpeg-b-plc两亲性嵌段共聚物。通过本发明制备方法制备而得的mpeg-b-plc两亲性嵌段共聚物分子量分布为1.2~1.4,具有分子量分布窄的优点,并且锡元素含量为5~40ppm,具有锡元素含量低的优点。采用jy/t015-1996电感耦合等离子体原子发射光谱方法测试上述实施例mpeg-b-plc两亲性嵌段共聚物中锡离子含量,并且测定分子量分布,测试结果如表1所示,本发明制备的mpeg-b-plc两亲性嵌段共聚物的锡离子含量低,有利于降低制剂毒副作用。表1mpeg-b-plc两亲性嵌段共聚物sn含量测试结果sn含量(单位ppm)分子量(mn)分子量分布实施例1359.94e31.39实施例263.09e31.21实施例3113.47e31.32本发明多西紫杉醇纳米制剂由多西紫杉醇和mpeg-b-plc两亲性嵌段共聚物组成。在具体实施例中,多西紫杉醇与mpeg-b-plc两亲性嵌段共聚物的质量比为1:3~100,优选质量比为1:10。本发明多西紫杉醇纳米制剂的制备方法,包括步骤:s1、将多西紫杉醇溶解于第一有机溶剂,得到第一溶液。在具体实施例中,该第一有机溶剂为无水乙醇,将多西紫杉醇粉末溶解于无水乙醇中,所得第一溶液中多西紫杉醇浓度为0.1~0.5g/l,优选浓度为0.2g/l。s2、将mpeg-b-plc两亲性嵌段共聚物溶解于第二有机溶剂,得到第二溶液。在具体实施例中,第二有机溶剂为乙腈,将mpeg-b-plc两亲性嵌段共聚物粉末溶解于乙腈中,所得第二溶液中mpeg-b-plc两亲性嵌段共聚物的浓度为0.3~5g/l,优选浓度为2g/l。s3、将脂肪酸溶解于第三有机溶剂,得到第三溶液。在具体实施例中,脂肪酸选自月桂酸、棕榈酸、油酸、亚油酸中任一种,第三有机溶剂为无水乙醇。所得第三溶液中,脂肪酸的浓度为浓度为0.1~2g/l,优选浓度为1g/l。s4、混合第一溶液、第二溶液和第三溶液,去除有机溶剂,获取混合物。在具体实施例中,将第一溶液、第二溶液、第三溶液按照如下体积比混合:1~100:1~100:1,优选25:25:1。在具体实施例中,将第一溶液、第二溶液和第三溶液于60℃水浴下搅拌混合30分钟,再在60℃温度下负压去除有机溶剂,获得紫杉醇和mpeg-b-plc两亲性嵌段共聚物的混合物。s5、水化所述混合物,用纳米过滤器过滤,获取滤液。在具体实施例中,用蒸馏水水化混合物,震动混合15~30分钟获得混合液,用200纳米过滤器过滤混合液,获得滤液。s6、冷冻所述滤液,获取多西紫杉醇纳米冰块。在具体实施例中,在-40℃条件下冷冻滤液,获取多西紫杉醇纳米冰块。s7、冷冻干燥所述多西紫杉醇纳米冰块,获取多西紫杉醇纳米制剂。在具体实施例中,将紫杉醇纳米冰块用冻干机干燥48~72小时,获取多西紫杉醇纳米制剂。实施例4多西紫杉醇纳米制剂的制备及注射液的制备本实施例中,多西紫杉醇纳米制剂的制备方法包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)0.5gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg月桂酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃,旋转混合30分钟;并在60℃温度下,负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg(2k)-b-plc(xk)两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得多西紫杉醇纳米制剂。本实施例制备的多西紫杉醇纳米制剂中,多西紫杉醇和mpeg-b-plc两亲性嵌段共聚物的质量比为1:10。使用本发明制备的多西紫杉醇醇纳米制剂时,采用生理盐水将其配置成注射液,注射剂配置方法如下:25ml0.9%生理盐水溶解上述多西紫杉醇纳米制剂,待获得透明液体,稳定性为24小时,可用于静脉注射。实施例5多西紫杉醇纳米制剂的制备及注射液的制备本实施例中,多西紫杉醇纳米制剂的制备方法包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)0.5gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg棕榈酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃旋转混合30分钟;并在60℃温度下,大约2小时,负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg(2k)-b-plc(xk)两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得紫杉醇纳米制剂。注射液配置:25ml0.9%生理盐水溶解本实施例多西紫杉醇纳米制剂,待获得透明液体,稳定性为5小时,可用于注射。实施例6多西紫杉醇纳米制剂的制备和注射液配制本实施例中,多西紫杉醇纳米制剂的制备包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)0.5gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg油酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃,旋转混合30分钟;并在60℃温度下,负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg(2k)-b-plc(xk)两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得多西紫杉醇纳米制剂。本实施例多西紫杉醇注射液配置方法如下:用25ml0.9%生理盐水溶解上述多西紫杉醇纳米饼,待获得透明液体,稳定性为5小时,可用于注射。实施例7多西紫杉醇纳米制剂的制备和注射液配制本实施例中,多西紫杉醇纳米制剂的制备包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)0.5gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg亚油酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃,旋转混合30分钟;并在60℃温度下,2小时负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg-b-plc两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得多西紫杉醇纳米制剂。本实施例多西紫杉醇注射液配置方法如下:用25ml0.9%生理盐水溶解上述多西紫杉醇纳米饼,待获得透明液体,稳定性为5小时,可用于注射。实施例8多西紫杉醇纳米制剂的制备和注射液配制本实施例中,多西紫杉醇纳米制剂的制备包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)0.15gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg亚油酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃,旋转混合30分钟;并在60℃温度下,2小时负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg-b-plc两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得多西紫杉醇纳米制剂。本实施例多西紫杉醇注射液配置方法如下:用25ml0.9%生理盐水溶解上述多西紫杉醇纳米饼,待获得透明液体,稳定性为4小时,可用于注射。实施例9多西紫杉醇纳米制剂的制备和注射液配制本实施例中,多西紫杉醇纳米制剂的制备包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)5gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg亚油酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃,旋转混合30分钟;并在60℃温度下,2小时负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg-b-plc两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得多西紫杉醇纳米制剂。本实施例多西紫杉醇注射液配置方法如下:用25ml0.9%生理盐水溶解上述多西紫杉醇纳米饼,待获得透明液体,稳定性为24小时,可用于注射。实施例10多西紫杉醇纳米制剂的制备和注射液配制本实施例中,多西紫杉醇纳米制剂的制备包括如下步骤:a)50mg多西紫杉醇粉末溶解于250ml无水乙醇溶剂中。b)2.5gmpeg(2k)-b-plc(xk)粉末溶解于250ml乙腈溶剂中。c)10mg亚油酸溶解于10ml无水乙醇中。d)混合上述三种溶液:在旋转蒸发仪的玻璃瓶中,水浴60℃,旋转混合30分钟;并在60℃温度下,2小时负压除去乙醇和乙腈溶剂,剩下多西紫杉醇和两亲聚合物混合物。e)用25ml蒸馏水水化多西紫杉醇和mpeg-b-plc两亲聚合物的混合物,水化震动时间为15~30分钟。f)水化后的多西紫杉醇纳米溶液用200纳米过滤器过滤,过滤液放置于玻璃瓶中。g)玻璃瓶中水化后的多西紫杉醇纳米溶液放置在-40℃环境下冷冻。h)冷冻后的多西紫杉醇纳米冰块用冻干机干燥48小时,获得多西紫杉醇纳米制剂。本实施例多西紫杉醇注射液配置方法如下:用25ml0.9%生理盐水溶解上述多西紫杉醇纳米饼,待获得透明液体,稳定性为24小时,可用于注射。实验例药效学试验采用动物实验检测本发明多西紫杉醇纳米制剂对肺原位移植瘤并全身转移瘤的治疗作用。(1)动物模型的制备和分组对nu/nu裸小鼠肺进行原位穿刺注射h460细胞,14天后,采集荷瘤裸鼠的生物发光信号,并以此为依据分为4组,每组10只荷瘤裸鼠,开始静脉注射给药,时间记为day1。上述分组包括对照组、paclitaxel(紫杉醇)治疗组、dtx-fatacid治疗组1、dtx-fatacid治疗组2。其中对照组小鼠未接受任何治疗;paclitaxel组小鼠接受taxol(泰素)静脉注射治疗,用药量为20mg每kg小鼠体重;dtx-fatacid治疗组1小鼠被静脉注射本发明多西紫杉醇纳米注射剂,用药量为20mg/kg;dtx-fatacid治疗组2中,小鼠同样接受本发明多西紫杉醇纳米注射剂,用药量为40mg/kg。图1所示为day1各组生物发光强度分布,图2所示为day1各组生物发光2d成像,可见在day1时,各组荷瘤裸鼠癌细胞h460数量及分布情况相近。(2)治疗效果分析本试验对荷瘤裸鼠的治疗方式为药物静脉注射,在day1、day3、day5、day7、day9分别进行给药。第一次给药后第8天,记为day8,采集各组荷瘤裸鼠的生物发光信号,结果如图3、图4所示。与对照组相比,paclitaxel治疗组生物发光较弱,说明其对肺原位移植瘤有一定的治疗效果,而两dtx-fatacid治疗组的生物发光强度更低,说明本发明多西紫杉醇纳米制剂能有效抑制肿瘤的快速发展。对照组在day4、day7分别有1只裸鼠死亡,paclitaxel治疗组出现了3只裸鼠死亡,两dtx-fatacid治疗组的荷瘤裸鼠均保持存活。观察发现,taxol注射后,小鼠立即出现毒性反应,如俯卧不动、呼吸急促、体温降低等,并且注射部位早期出现部刺激症状、晚期出现坏死脱落,反映taxol显示出急性毒性作用,而本发明多西紫杉醇纳米制剂则未出现毒副作用。第一次给药后第15天,记为day15,采集各组荷瘤裸鼠的生物发光信号,图5展示各组荷瘤裸鼠生物发光强度,图6是各组荷瘤裸鼠的生物发光2d成像。图5中空心符号代表已死亡的动物数据,图6中方框表示该裸鼠已死亡,标记时间为死亡时间,可见day15时,对照组全部裸鼠死完,paclitaxel组仅存活3只荷瘤裸鼠,dtx-fatacid治疗组1出现1只裸鼠死完。第一次给药后第22天,记为day22,采集各组荷瘤裸鼠的生物发光信号,图7、图8分别展示day22各组荷瘤裸鼠生物发光强度、各组荷瘤裸鼠的生物发光2d成像。day22时,paclitaxel组仅存活1只荷瘤裸鼠,dtx-fatacid治疗组的裸鼠存活状况较好,dtx-fatacid治疗组1有两只裸鼠死完,dtx-fatacid治疗组2全部裸鼠存活。从图7生物发光强度对比可见,两dtx-fatacid治疗组的生物发光强度明显低于对照组和paclitaxel治疗组,说明本发明多西紫杉醇纳米制剂有良好的抑制肿瘤效果。图9所示为各组荷瘤裸鼠随着病程进展生物发光强度增长倍数的比较,反映在不同时间control组、paclitaxel组、dtx-fatacid组小鼠肿瘤大小的变化。根据结果显示,随着肿瘤的发展,对照组小鼠的生物发光强度最终增长为day1强度的24.8倍,paclitaxel组为15.9倍,dtx-fatacid治疗组1(即给药量为20mg/kg组)为1.6倍,dtx-fatacid治疗组2(即给药量为40mg/kg组)为1.1倍。发光强度的增长反映肿瘤的发展,图9的结果显示,说明相比于paclitaxel组,本发明的多西紫杉醇纳米制剂具有更好的抑制疾病进程的效果,给药量与抑制肿瘤的效果呈正相关。由于疾病恶性,肺原位移植瘤的小鼠模型会出现体重下降的情况,图10展示各组荷瘤裸鼠体重增长倍数的比较。对照组中,荷瘤裸鼠出现快速的体重下降;taxol虽然有一定的抑瘤效果,但是由于其毒副作用,该组小鼠体重出现明显下降,并保持在较低水平;两dtx-fatacid治疗组,荷瘤裸鼠体重变化较小,反映肿瘤得到有效控制,并且本发明多西紫杉醇自己无明显毒副作用。统计各组荷瘤裸鼠的生存期,结果如表2所示,dtx-fatacid治疗组2的中位生存期为41.5天,dtx-fatacid治疗组1的中位生存期为28.5天,约为对照组荷瘤裸鼠中位生存期(10天)的4倍和3倍;约为paclitaxel(20mg/kg)组荷瘤裸鼠中位生存期(14天)的3倍和2倍。可见本发明多西紫杉醇纳米制剂的治疗,能有效控制肺原位移植瘤的病程发展,延长荷瘤裸鼠的生存期。表2各组荷瘤裸鼠生存期分析本实验例,还通过3d精准成像来观察肿瘤转移情况,对照组肿瘤全身转移的发生率为10/10,paclitaxel治疗组肿瘤全身转移发生率为7/8,dtx-fatacid治疗组1肿瘤全身转移发生率为3/10,而给药量更高的dtx-fatacid治疗组2没有小鼠出现全身转移的情况,说明本发明多西紫杉醇制剂有效抑制肿瘤的全身转移。根据药效学试验结果,本发明多西紫杉醇纳米制剂更够有效延缓肿瘤的进展以及控制全身转移,提高荷瘤小鼠生存期,并且无明显毒副作用。显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1