促性激素释放激素受体拮抗剂及其用途的制作方法
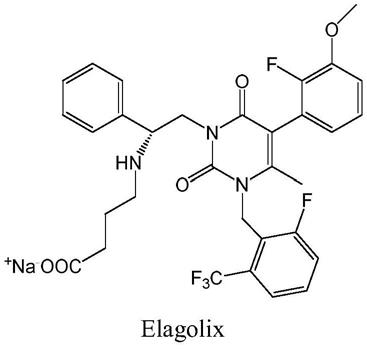
6-[trifluoromethyl]-benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2h-pyrimidin-1-yl]-1-phenylethylamino}butyrate(elagolix),a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor,j.med.chem.2008,51,7478
–
7485)。但采用放射性标记的elagolix口服给予大鼠,通过胆汁排泄的总放射性接近50%,显示elagolix可能存在肝脏首过效应,大部分药物未能进入循环发挥效应。从而导致临床治疗剂量较高(elagolix推荐临床剂量为150mg,qd或者200mg,bid)。而且大鼠口服后总放射性主要局限性分布于胃肠道和肝脏。elagolix在人体也显示较高的表观分布容积(1674l),均提示高度组织分布特征。过高的肝脏分布,可导致elagolix肝毒性风险(肝脏转氨酶升高)以及对肝药酶的影响(elagolix是cyp3a、p-gp和oatp1b1的底物,也是p-gp的抑制剂,对p450(cyp)3a具有弱到中等程度诱导作用)(fda,orilissa(elagolix),210450orig1s000multid,2018)。
[0009][0010]
虽然在该领域已进行了有意义的研究,但仍需要药性更为理想的有效的小分子gnrh受体拮抗剂。同时也需要含有此gnrh受体拮抗剂的药物组合物,以及利用其来治疗诸如性激素相关的疾病状态的方法。
技术实现要素:
[0011]
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物及其制备方法和用途,用于解决现有技术中的问题。
[0012]
为实现上述目的及其他相关目的,本发明一方面提供一种化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物,所述化合物的化学结构式如式i所示:
[0013][0014]
在本发明一些实施方式中,所述异构体选自对映异构体、非对映异构体、顺反异构体或立体异构体。
[0015]
本发明另一方面提供上述的化合物的制备方法,包括:将式1-12化合物进行水解,制备获得式i化合物,反应方程式如下:
[0016][0017]
在本发明一些实施方式中,所述水解反应通常可以在碱存在的条件下进行。
[0018]
本发明另一方面提供上述的化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物在制备药物中的用途。
[0019]
在本发明一些实施方式中,所述药物选自促性腺激素释放激素受体拮抗剂。
[0020]
在本发明一些实施方式中,所述药物选自用于治疗性腺激素相关的疾病的药物。
[0021]
在本发明一些实施方式中,所述性腺激素相关的疾病选自子宫内膜异位症、闭经、月经不调、子宫肌瘤、子宫纤维瘤、多囊卵巢病、子宫内膜异位症、子宫平滑肌瘤、红斑狼疮、多毛症、性早熟、矮小症、痤疮、脱发、性腺激素依赖性肿瘤、产生促性腺激素的垂体腺瘤、睡眠呼吸暂停、肠易激综合征、经前期综合征、良性前列腺增生、避孕和不育、阿尔茨海默病。
[0022]
在本发明一些实施方式中,所述性腺激素依赖性肿瘤选自前列腺癌、子宫癌、乳腺癌、卵巢癌、垂体促性腺细胞腺瘤。
[0023]
本发明另一方面提供一种药物组合物,包括上述的化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物。
附图说明
[0024]
图1显示为本发明实施例3中sd大鼠静脉注射g201/elagolix的药时曲线示意图。
[0025]
图2显示为本发明实施例3中sd大鼠口服给予g201/elagolix的药时曲线示意图。
[0026]
图3显示为本发明实施例4中icr小鼠静脉及口服给予g201的药时曲线示意图。
具体实施方式
[0027]
为了使本发明的发明目的、技术方案和有益技术效果更加清晰,以下结合实施例对本发明进行进一步详细说明,熟悉此技术的人士可由本说明书所揭露的内容容易地了解本技术发明的其他优点及功效。
[0028]
本发明发明人经过大量实践研究,通过对elagolix结构修饰,意外发现一类新的氟取代嘧啶二酮类化合物,所述化合物相较于现有技术中常用的elagolix等具有更佳的生物活性和更理想的药代动力学特性,从而提供了一种新的可以作为促性腺激素释放激素(gnrh)受体拮抗剂的化合物,在此基础上完成了本发明。
[0029]
本发明第一方面提供一种化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物,所述化合物的化学结构式如式i所示:
[0030][0031]
本发明中,术语“盐”应当被理解为由本发明使用的任何形式的活性化合物,其中所述化合物可以为离子形式或带电荷或被偶联到反离子(阳离子或阴离子)或在溶液中。这个定义还可以包括活性分子与其它分子和离子的季铵盐和络合物,特别是通过离子相互作用的络合物。该定义尤其包括生理上可接受的盐,该术语可以被理解为与“药理学上可接受的盐”等同。
[0032]
本发明中,术语“药学上可接受的盐”通常指当以适当的方式用于治疗时(特别是在人类和/或哺乳动物中应用或使用时)在生理学上可耐受的任何盐(通常来说,这意味着它是无毒的,特别是作为抗衡离子的结果是无毒的)。这些生理上可接受的盐可以是与阳离子或碱形成的,并且在本发明的上下文中,尤其是在人类和/或哺乳动物中施用时,它们应该被理解为由按照本发明所提供的至少一种化合物,通常为酸(去质子化的),如阴离子和至少一种生理学上耐受的阳离子(优选无机阳离子)形成的盐。在本发明的上下文中,具体地可以包括与碱金属和碱土金属形成的盐、以及与铵阳离子(nh
4+
)形成的盐,具体可以是包括但不限于与(单)或(二)钠、(单)或(二)钾、镁或钙形成的盐。这些生理上可接受的盐也可以是与阴离子或酸形成的,并且在本发明的上下文中,特别是在人类和/或哺乳动物中施用时,它们应该被理解为由按照本发明所提供的至少一种化合物,通常质子化的(例如在氮上),如阳离子和至少一种生理上可耐受的阴离子形成的盐。在本发明的上下文中,具体地可以包括由生理上可耐受的酸形成的盐,即特定的活性化合物与生理上可耐受的有机或无机酸形成的盐,具体可以是包括但不限于与盐酸、氢溴酸、硫酸、甲磺酸、甲酸、乙酸、草酸、琥珀酸、苹果酸、酒石酸、扁桃酸、富马酸、乳酸或柠檬酸形成的盐。示例性的式i化合物的药学上可接受的盐的化学结构式如下:
[0033][0034]
由上述式i表示的本发明化合物可以包括取决于存在的手性中心的对映体或取决于存在的双键的异构体(例如z,e)。单一异构体、对映异构体、非对映异构体或顺反异构体和它们的混合物均落入本发明的范围之内。
[0035]
本发明中术语“前药”以其最广泛的意义使用,并且包括在体内可以转化为本发明的化合物的那些衍生物。制备指定的起作用化合物的前药的方法对于本领域技术人员来说应该是已知的,例如,可以参阅如krogsgaard-larsen等人,“药物设计和发现教科书”(textbook of drug design and discovery)泰勒弗朗西斯出版社taylor&francis(2002年4月)中所公开的相关内容。
[0036]
本发明中,术语“溶剂化物”通常指任何形式的根据本发明的活性化合物通过非共价键与另一分子(通常为极性溶剂)相结合,所获得的物质,具体可以是包括但不限于水化物和醇化物,例如甲醇化物。
[0037]
本发明第二方面提供本发明第一方面所提供的化合物的制备方法,包括:将式1-12化合物进行水解,制备获得式i化合物,反应方程式如下:
[0038][0039]
本发明所提供的制备方法中,所述水解反应通常可以在碱存在的条件下进行。本领域技术人员可选用合适种类和用量的碱,用于上述水解反应,例如,所述碱可以是碱金属的氢氧化物等,更具体可以是氢氧化锂等,再例如,所述碱的用量相对于式1-12化合物通常是基本等量或者过量的,具体的,式1-12化合物与碱的摩尔比可以为1:1.4~1.6。
[0040]
本发明所提供的制备方法中,反应通常可以在室温至反应溶剂沸点的条件下进行,优选可以在室温下进行。本领域技术人员可根据反应进程适当调整水解反应的反应时间,监测反应进程的方法对于本领域技术人员来说应该是已知的,例如可以是色谱法、核磁共振法等分析方法,具体的反应时间可以是例如0.5~1小时、1~1.5小时、1.5~2小时、2~3小时、3~4小时或更长的反应时间。
[0041]
本发明所提供的制备方法中,反应通常在溶剂存在的条件下进行,所述溶剂通常
可以是反应原料的良溶剂、且需要包括水,从而可以充分分散反应原料并保证反应的顺利进行。合适的反应溶剂的种类和用量对于本领域技术人员来说应该是已知的,例如,反应溶剂可以包括水,还可以包括醚类溶剂等,具体可以是四氢呋喃等。
[0042]
本发明所提供的制备方法中,本领域技术人员可选择合适的方法对反应产物进行后处理,例如,可以包括:高效色谱制备、冻干等。
[0043]
本发明第三方面提供本发明第一方面所提供的化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物在制备药物中的用途。本发明的化合物能有效抑制gnrh受体,从而可以作为促性腺激素释放激素受体拮抗剂,并可以进一步被用于治疗性激素相关疾病。本发明所提供的促性腺激素释放激素受体拮抗剂可应用于广泛的治疗用途,并可用来治疗男性和女性以及一般哺乳动物(本发明也称为“个体”)的各种性激素相关的疾病状态。所述性激素相关疾病具体可以是例如子宫内膜异位症、闭经、月经不调、子宫肌瘤、子宫纤维瘤、多囊卵巢病、子宫内膜异位症、子宫平滑肌瘤、红斑狼疮、多毛症、性早熟、矮小症、痤疮、脱发、性腺激素依赖性肿瘤(例如,前列腺癌、子宫癌、乳腺癌、卵巢癌、垂体促性腺细胞腺瘤等)、产生促性腺激素的垂体腺瘤、睡眠呼吸暂停、肠易激综合征、经前期综合征、良性前列腺增生、避孕和不育、阿尔茨海默病等。
[0044]
本发明第四方面提供一种药物组合物,包括本发明第一方面所提供的化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物,所述药物组合物还可以包括至少一种药学上可接受的载体。
[0045]
本发明中,所述组合物可以包括一种或多种药学上可接受的载体,其通常指用于治疗剂给药的载体,它们本身不诱导产生对接受该组合物的个体有害的抗体,且给药后没有过分的毒性。这些载体是本领域技术人员所熟知的,例如,在remington’s pharmaceutical sciences(mack pub.co.,n.j.1991)中公开了关于药学上可接受的载体的相关内容。具体来说,所述载体可以是包括但不限于盐水、缓冲液、葡萄糖、水、甘油、乙醇、佐剂等中的一种或多种的组合。
[0046]
本发明所提供的药物组合物中,所述化合物可以是单一有效成分,也可以与其他活性组分进行组合,构成联合制剂。所述其他活性组分可以是其他各种可以用于治疗性腺激素相关的疾病的药物。组合物中活性组分的含量通常为安全有效量,所述安全有效量对于本领域技术人员来说应该是可以调整的,例如,所述化合物和药物组合物的活性成分的施用量通常依赖于患者的体重、应用的类型、疾病的病情和严重程度,例如,作为活性成分的所述化合物的施用量通常可以为0.1~10mg/kg/day、0.1~0.5mg/kg/day、0.5~1mg/kg/day、1~2mg/kg/day、2~3mg/kg/day、3~4mg/kg/day、4~5mg/kg/day、5~6mg/kg/day、6~8mg/kg/day、8~10mg/kg/day,更优为0.5~5mg/kg/day。
[0047]
本发明所提供的化合物可以适应于任何形式的给药方式,可以是口服或胃肠外给药,例如,可以是经肺、经鼻、经直肠和/或静脉注射,更具体可以是真皮内、皮下、肌内、关节内、腹膜内、肺部、口腔、舌下含服、经鼻、经皮、阴道、口服或胃肠外给药。本领域技术人员可根据给药方式,选择合适的制剂形式,例如,适合于口服给药的制剂形式可以是包括但不限于丸剂、片剂、咀嚼剂、胶囊剂、颗粒剂、滴剂或糖浆等,再例如,适合于胃肠外给药的制剂形式可以是包括但不限于溶液、悬浮液、可复水的干制剂或喷雾剂等,再例如,适合于直肠给药的通常可以是栓剂。
[0048]
本发明第五方面提供一种治疗方法包括:向个体施用治疗有效量的本发明第一方面所提供的化合物、或本发明第四方面所提供的药物组合物。
[0049]
本发明中,“个体”通常包括人类、非人类的灵长类,如哺乳动物、狗、猫、马、羊、猪、牛等,其可因利用所述制剂、试剂盒或联合制剂进行治疗而获益。
[0050]
本发明中,“治疗有效量”通常指一用量在经过适当的给药期间后,能够达到治疗如上所列出的疾病的效果。
[0051]
本发明提供的化合物在钙流检测实验中作为gnrhr拮抗剂,活性与elagolix相当或更优;在药代动力学实验中,本发明所提供的化合物绝对生物利用度明显高于elagolix;在血浆蛋白结合试验中,本发明所提供的化合物与sd大鼠血浆及健康人体血浆蛋白结合率略高于elagolix,且蛋白结合无药物浓度依赖关系。可见,本技术所提供的化合物或其药学上可接受的盐、异构体、前药、多晶型物或溶剂化物相较于现有技术中其他同类药物具有更佳的生物活性和更理想的药代动力学特性,具有良好的产业化前景。
[0052]
下面通过实施例对本技术的发明予以进一步说明,但并不因此而限制本技术的范围。
[0053]
实施例1
[0054]
实施例中化合物g201的具体制备路线如下:
[0055][0056]
在三口瓶中加入化合物1-1(10g,72.4mmol)、吡啶(200ml)和二氧化硒(16g,144.8mmol)。氮气保护下100℃反应2小时。反应完毕后抽滤,滤液旋干,然后加入1n盐酸,用乙酸乙酯萃取。有机相用饱和食盐水洗,无水硫酸钠干燥,抽滤,旋干得类白色固体1-2(12g)。
[0057]
在三口瓶中加入化合物1-2(12g,71.4mmol)和甲醇(200ml)。0℃下滴加氯化亚砜(17g,142.8mmol),室温搅拌过夜。反应完毕后旋干甲醇,加入饱和碳酸氢钠水溶液,用乙酸乙酯萃取。有机相用饱和食盐水洗,无水硫酸钠干燥,抽滤,旋干得浅黄色油状物1-3(9g,49.4mmol)。
[0058]
在三口瓶中加入化合物1-3(4g,20.4mmol)、无水四氢呋喃(60ml)和(r)-叔丁基亚磺酰胺(2.47g,20.4mmol)。氮气保护下加入钛酸四异丙酯(11.6g,40.8mmol),回流搅拌6小时。反应完毕后加水淬灭反应,加乙酸乙酯稀释,然后抽滤。滤液分液,水相用乙酸乙酯萃
取。合并有机相,用饱和食盐水洗,无水硫酸钠干燥,抽滤,旋干得黄色油状物1-4(6g,19.2mmol)。
[0059]
在三口瓶中加入化合物1-4(6g,19.2mmol)和甲醇(60ml)。0℃下分批加入硼氢化钠(1.1g,28.7mmol),室温反应3小时。反应完毕后加水淬灭反应,用乙酸乙酯萃取。有机相用饱和食盐水洗,无水硫酸钠干燥,抽滤,旋干,柱层析纯化(石油醚/乙酸乙酯=10:1-5:1)得黄色油状物1-5(3g,9.5mmol)。
[0060]
在三口瓶中加入化合物1-5(3g,9.5mmol)和无水四氢呋喃(40ml)。0℃下分批加入氢化铝锂(0.43g,11.4mmol),室温下反应2小时。反应完毕后加水淬灭反应,用乙酸乙酯萃取。有机相用饱和食盐水洗,无水硫酸钠干燥,抽滤,旋干,柱层析纯化(石油醚/乙酸乙酯=10:1-3:1)得黄色油状物物1-6(1.5g,5.8mmol)。
[0061]
在单口瓶中加入化合物1-6(1.5g,5.8mmol)、乙酸乙酯(10ml)和盐酸气/乙酸乙酯(10ml)。室温下反应1小时。反应完毕后旋干得黄色固体1-7(1.5g),直接投下一步。
[0062]
单口瓶中加入化合物1-7(1.5g)、二氯甲烷(20ml)、三乙胺(2.9g,28.9mmol)和二碳酸二叔丁酯(1.5g,6.9mmol)。室温反应16小时。反应完毕后旋干,柱层析纯化(石油醚/乙酸乙酯=15:1-10:1)得黄色固体1-8(1.3g,5.1mmol)。
[0063]
三口瓶中加入化合物1-8(1.3g,5.1mmol)、化合物1-a(1.9g,5.1mmol)、三苯基膦(2.0g,7.6mmol)、无水四氢呋喃(30ml)和偶氮二甲酸二乙酯(1.5g,7.6mmol)。回流反应16小时。反应完毕后旋干,柱层析纯化(石油醚/乙酸乙酯=10:1-5:1)得白色固体1-9(2.1g,3.4mmol)。
[0064]
三口瓶中加入化合物1-9(2.1g,3.4mmol)、化合物1-b(0.7g,4.1mmol)、碳酸钠(1.4g,13.6mmol)、二氧六环(27ml)、水(9ml)和四(三苯基膦)钯(0.4g,0.34mmol)。氮气保护下90℃反应16小时。反应完毕后抽滤,滤液旋干,柱层析纯化(石油醚/乙酸乙酯=15:1-5:1)得浅黄色固体1-10(1.35g,2.0mmol)。
[0065]
单口瓶中加入化合物1-10(250mg,0.38mmol)、二氯甲烷(2ml)和三氟乙酸(2ml),室温下反应1小时。反应完毕后旋干,得黄色固体1-11(210mg)。
[0066]
单口瓶中加入粗品化合物1-11(210mg)、乙腈(3ml)、化合物1-c(220mg,1.13mmol)和碳酸钾(156mg,1.13mmol),80℃反应16小时。反应完毕后旋干,制备薄层色谱(石油醚/乙酸乙酯=1.5/1),得无色油状物1-12(210mg,0.31mmol)。
[0067]
单口瓶中加入化合物1-12(210mg,0.31mmol)、四氢呋喃(3ml)、水(2ml)和氢氧化锂(20mg,0.47mmol),室温下反应2小时。反应完毕后旋干,高压制备(乙腈10-55/7分钟),冻干得白色固体g201(20mg)。1hnmr cd3odδ:7.62-7.64(m,1h),7.39-7.57(m,4h),7.11-7.23(m,4h),6.65-6.80(m,1h),5.38-5.49(m,2h),4.81(q,1h),4.46-4.53(m,2h),3.90(s,3h),2.81-2.84(m,2h),2.37-2.40(m,2h),2.12(d,3h),1.79-1.83(m,2h).lc-ms:m/z=649.8(m+1).,lcms纯度>98%。
[0068]
实施例2
[0069]
钙流检测:
[0070]
使用flipr钙流检测试剂盒(calcium 4assay kit)测量重组人促性腺激素释放激素受体(gnrhr)稳定细胞系cho-k1/gnrhr/gα15细胞内钙变化。gnrhr是一种g蛋白偶联受体(gpcrs),gpcrs信号通过gq通路使细胞内钙释放,因此,通过钙离子敏感荧光探针检测细胞
内钙释放,即可检测通过gq途径进行信号转导的gnrhr的功能变化。实验步骤如下:
[0071]
1.用f12+10%fbs培养基,按照10000cells/well、20ul/well,将细胞种于384孔板(corning,cat#:3764)中,培养过夜(18h)。
[0072]
2.将丙磺舒储液(500mm)和calcium 4储液按1:100配制成染料工作液。用测定缓冲液(assay buffer)稀释供试品g201和elagolix(2mm in dmso):取5μl 2mm储存液稀释至200ul(50um),此即第1个浓度的5x样品工作液。取20ul的第1样品工作液,稀释至200ul,混匀后即得第2浓度样品工作液。依次共准备8个梯度稀释的样品工作液。向培养过夜的细胞中依次加入20ul/well染料工作液和10ul/well样品工作液后,继续在细胞培养箱中孵育45min,然后室温下避光平衡15min。
[0073]
3.5x ec80激动剂工作液配置:取5μl 1mm buserelin储存液稀释至200ul(25um),取71ul 25um的buserelin工作液,用assay buffer稀释至10ml(0.179um)。
[0074]
4.对应于384孔板中细胞的位置,按40ul/well将激动剂工作液加入进样板,行flipr检测。
[0075]
试验结果:g201可剂量依赖性抑制cho-k1/gnrhr/gα15细胞内钙释放,ec
50
为37.59nm,elagolix为45.73nm。表明g201为gnrhr拮抗剂,活性与elagolix相当或更优。
[0076]
实施例3
[0077]
大鼠药代动力学试验:
[0078]
雌性sd大鼠12只,分为4组,3只/组,分别静脉注射和灌胃给予g201和elagolix sodium(g201溶于含5%n,n-二甲基乙酰胺及5%聚氧乙烯蓖麻油el的生理盐水,elagolix sodium直接溶于生理盐水),剂量分别为25mg/kg和50mg/kg。给药前禁食过夜,给药后4h恢复给食,整个试验期间自由饮水。在给药前及给药后5、15、30min,1、2、4、6、8、24h,眼眶后静脉丛采血,采血体积约0.2ml,全血样品于1h内4000rpm离心10min,分离上层血浆且1h内放置冰箱-20℃保存至分析。血浆样品1:8经甲醇蛋白沉淀后,通过lc-ms/ms方法检测g201和elagolix血药浓度,应用das3.2.7软件计算主要药代动力学参数。
[0079]
结果显示:g201静脉注射给药平均cl为1.62l/h/kg,平均t
1/2
为0.79h,平均vz为1.82l/kg。而elagolix sodium平均cl和vz分别为2.48l/h/kg和8.46l/kg。g201表观分布容积明显低于elagolix,显示组织分布降低。
[0080]
大鼠口服给予g201后吸收较快,t
max
为0.42h,,平均auc
0-∞
为16351.7h*ng/ml,相当于elagolix sodium的三倍。g201口服的绝对生物利用度为51.92%,明显高于elagolix sodium(23.65%)。g201/elagolix静脉注射给药药时曲线见图1,口服给药药时曲线见图2,药代动力学参数见表1。
[0081]
表1.g201和elagolix在sd大鼠的药代动力学参数(平均值,n=3)
[0082][0083]
auc:血药浓度-时间曲线下面积;t1/2z:消除半衰期;tmax:达峰时间;vz:表观分布容积;clz:清除率;cmax:血药峰浓度;f:绝对生物利用度,下同。
[0084]
实施例4
[0085]
小鼠药代动力学试验:
[0086]
雌性icr小鼠10只分为两组,每组5只,分别静脉注射和口服灌胃给予g201,剂量为10mg/kg,给药后0.083、0.25、0.5、1、2、4、6、8h眼眶后静脉丛采血,采血体积约0.1ml,血样于1h内4000rpm离心10min,分离上层血浆且1h内放置冰箱-80℃保存,待测。采血前受试动物禁食至少12h,不禁水。采血过程中禁食禁水,给药2h后自由饮食饮水。通过lc-ms/ms方法检测g201在icr小鼠体内血药浓度,应用das 3.2.7药代动力学软件计算主要药代动力学参数,包括:auc
0-t
、auc
0-∞
、c
max
、t
1/2
等。
[0087]
结果显示,g201在小鼠静脉注射给药平均cl为3.18l/h/kg,平均t
1/2
为0.26h,平均vz为1.17l/kg。小鼠口服给予g201后迅速吸收,t
max
为0.27h,平均auc
0-∞
为1266.3h*ng/ml,绝对生物利用度为38.9%,明显高于elagolix(根据文献报道,elagolix在小鼠10mg/kg剂量下的绝对生物利用度为10%)。icr小鼠静脉及口服给予g201的药时曲线见图3,药代动力学参数见表2。
[0088]
表2.g201在icr小鼠口服和静脉给药后的药代动力学参数(平均值,n=10)
[0089][0090]
实施例5
[0091]
血浆蛋白结合试验:
[0092]
取预孵育过的sd大鼠(雌雄各5只)或健康人体血浆,分别与含有不同浓度的受试物(g201(1、10、100μm)和elagolix(10μm))及对照品(华法林钠(10μm))的工作液充分混合后,取50μl含药血浆样品加入等体积空白缓冲液作为未过滤样品,另转移350μl至超滤装置
中(超滤管内管),37℃离心(10000g
×
3分钟),离心结束后向已取出的外管样品(滤液样品)中加入50μl空白血浆,同样向已取出的内管样品(过滤剩余样品)中加入等体积的空白缓冲液。混合2分钟后,加入内标溶液。所有样品涡旋10分钟,离心沉淀蛋白,取上清液,采用lc-ms/ms方法测定受试物及对照品的浓度,计算游离百分数(游离百分数=(滤液样品药物浓度/未过滤血浆药物浓度)
×
100%)和结合百分数(结合百分数=100%-游离百分数)。回收率(%)=(滤液样品药物浓度
×
体积+过滤剩余样品药物浓度
×
体积)/未过滤血浆药物浓度
×
总体积
×
100。
[0093]
结果显示,elagolix 10μm药物浓度sd大鼠血浆及健康人体血浆蛋白结合率分别为87.71%、87.25%,与文献报道一致。g201与sd大鼠血浆及健康人体血浆蛋白结合率分别为89.79%、89.94%,略高于elagolix,在1-100μm范围蛋白结合无药物浓度依赖关系。
[0094]
表3.g201和elagolix与大鼠和人血浆蛋白结合率
[0095][0096]
综上所述,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
[0097]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1