一种发光材料、延迟荧光体及有机发光元件的制作方法
本发明属于发光材料领域,具体明涉及一种发光材料、延迟荧光体及使用其制备的有机发光元件。
背景技术:
自1987年第一篇关于有机电致发光二极管(organiclightemittingdiodes,oleds)的研究发表以来,oled的性能取得了巨大的提高,其中,高性能发光材料的开发最为重要。
有机发光材料的发光机理已经发展到热活性延迟荧光(tadf)材料。tadf材料通过三线态激子的反隙间穿越实现100%的内量子发光效率,并且,以其高效、无需贵金属、低成本等受到了关注。然而目前已发现的tadf材料较少,性能也有待提高,新型的可用于oled器件的tadf材料仍然亟待开发。
技术实现要素:
为了获得更多种类、更高性能的tadf材料,本发明的目的在于提供一种发光材料,延迟荧光体及使用其制备的有机发光元件。
本发明提供了一种发光材料,其包含通式(1)所表示的化合物:
通式(1)
上式中,r1~r10各自独立地表示氢原子或取代基;ar表示经取代或未经取代的芳香环或杂芳环;a为包括受体基团的基团,所述基团可被取代基取代。
作为该r1~r10表示的取代基,例如可列举:羟基、卤素原子、碳数1~20的烷基、碳数1~20的烷氧基、碳数1~20的烷硫基、碳数6~40的芳基、碳数3~40的杂芳基、碳数2~10的烯基、碳数2~10的炔基、碳数1~10的卤代烷基、碳数3~20的三烷基硅烷基、碳数4~20的三烷基硅烷基烷基、碳数5~20的三烷基硅烷基烯基、碳数5~20的三烷基硅烷基炔基等。这些具体例中,可进一步经取代基取代者也可被取代。更优选的取代基为碳数1~20的经取代或未经取代的烷基、碳数1~20的烷氧基、碳数6~40的经取代或未经取代的芳基、碳数3~40的经取代或未经取代的杂芳基。进而优选的取代基为碳数1~10的经取代或未经取代的烷基、碳数1~10的经取代或未经取代的烷氧基、碳数6~15的经取代或未经取代的芳基、碳数3~12的经取代或未经取代的杂芳基。
通式(1)中,ar表示经取代或未经取代的芳香环、经取代或未经取代的杂芳环、或这些中的任意两个连结而成的连结基。所述这些中的任意两个连结而成的连结基包含两个相同芳香环键结而成的基团、两个互不相同的芳香环键结而成的基团、两个相同杂芳环键结而成的基团、两个互不相同的杂芳环键结而成的基团、芳香环与杂芳环键结而成的基团。
如上所述,ar所可采取的芳香环可为单环的芳香环,也可为缩合环的芳香环。作为所述芳香环,优选的是苯环、萘环,更优选的是苯环。作为具体例,可列举1,2-亚苯基、1,3-亚苯基、1,4-亚苯基、1,2-亚萘基、1,3-亚萘基、1,4-亚萘基、1,5-亚萘基、1,8-亚萘基,优选的是1,4-亚苯基、1,4-亚萘基。ar所可取的杂芳环可为单环的杂芳环也可为缩合环的杂芳环。作为所述杂芳环,优选的是含有氮原子作为环骨架构成原子,氮原子的个数优选的是1~4个,更优选的是1~3个。缩合环的杂芳环也包含苯环及杂环的缩合环。作为构成杂芳环的环结构的例子,可列举吡啶环、哒嗪环、嘧啶环、三嗪环、三唑环、苯并三唑环,优选的是吡啶环。
ar所可取的芳香环及杂芳环的连结位置并无特别限定,例如在ar的情况下,其连结位置可为邻位、间位、对位中的任一个。
优选的,ar为苯环或联苯。
或者,所述通式(1)的ar为萘环。
优选的,所述通式(1)中的a可选自下述基团:
上述基团可被取代基取代,所述取代基可为羟基、卤素原子、碳数1~20的烷基、碳数1~20的烷氧基、碳数1~20的烷硫基、碳数6~40的芳基、碳数3~40的杂芳基、碳数2~10的烯基、碳数2~10的炔基、碳数1~10的卤代烷基、碳数3~20的三烷基硅烷基、碳数4~20的三烷基硅烷基烷基、碳数5~20的三烷基硅烷基烯基、碳数5~20的三烷基硅烷基炔基等。
优选的,上述发光材料,其中所述包括受体基团的基团a为下述通式(2)所表示的基团,
通式(2)
其中,r21~r210各自独立地表示氢原子或取代基;a2为受体基团,所述受体基团可被取代基取代;ar2为表示经取代或未经取代的芳香环或杂芳环。
其中,取代基r21~r210优选的范围可为上述的r1~r10取代基的举例;
ar2优选的范围可为上述的ar的举例。
优选的,所述通式(2)的ar2为苯环或联苯。
优选的,所述通式(2)中的受体基团a2可选子下述基团:
上述基团可被取代基取代,所述取代基可为羟基、卤素原子、碳数1~20的烷基、碳数1~20的烷氧基、碳数1~20的烷硫基、碳数6~40的芳基、碳数3~40的杂芳基、碳数2~10的烯基、碳数2~10的炔基、碳数1~10的卤代烷基、碳数3~20的三烷基硅烷基、碳数4~20的三烷基硅烷基烷基、碳数5~20的三烷基硅烷基烯基、碳数5~20的三烷基硅烷基炔基等。
本发明还提供了一种延迟荧光体,其包含上述的化合物。
本发明还包块一种有机发光元件,在基板上具有包含所述的发光材料的发光层。
进一步的,所述的有机发光元件,其放射延迟荧光。
同现有技术相比,本发明的有益效果体现在:
本发明采用新型的包含苯基膦基团的热活性延迟荧光材料,证明了苯基膦基团可以作为热活性延迟荧光分子中的给电子基团,通过与吸电子受体基团配合,能够实现热活性延迟荧光发光,该体系的分子能够覆盖较大的波长范围,具有较高的效率,是一类优秀的热活性延迟荧光材料。
附图说明
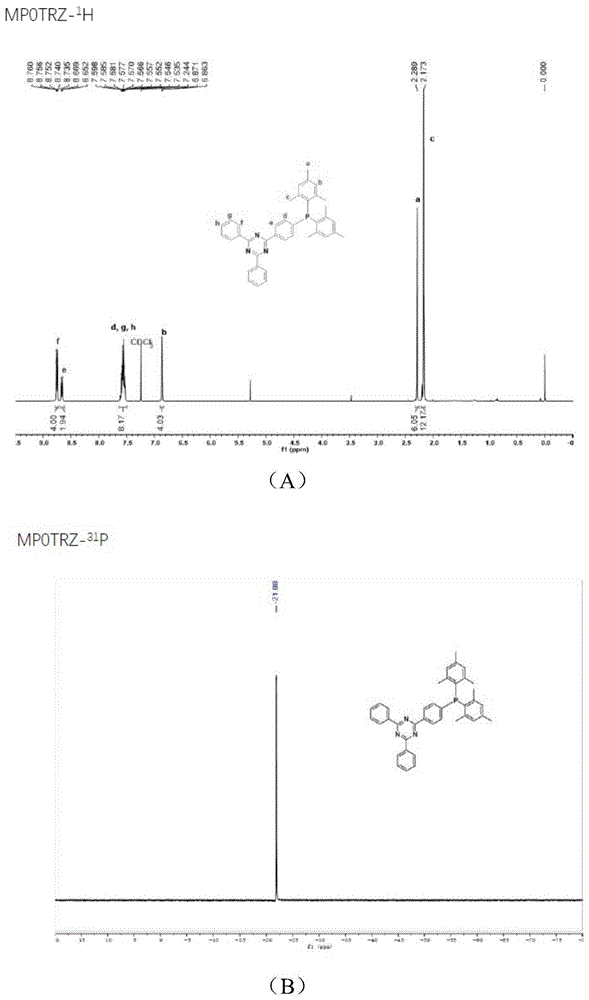
图1为实施例1得到的化合物mp0trz的核磁图谱;
图2为实施例1得到的化合物mp2trz的核磁图谱;
图3为实施例1得到的化合物iprp2trz的核磁图谱;
图4为实施例1得到的化合物mp0phtrz的核磁图谱;
图5为实施例1得到的化合物mp2phtrz的核磁图谱;
图6为实施例1得到的化合物iprp2phtrz的核磁图谱;
图7为实施例1得到的化合物在甲苯溶液中的光致发光光谱;
图8为实施例1得到的化合物在mcp薄膜中的光致发光光谱;
图9为实施例1得到的化合物的瞬态光致发光光谱衰减曲线;
图10为实施例2得到的器件的电压-电流密度-亮度曲线;
图11为实施例2得到的器件的电致发光光谱;
图12为实施例2得到器件的电流密度-外量子效率曲线。
具体实施方式
以下对本发明的内容进行详细说明。以下所记载的构成要件的说明有时是基于本发明的代表性实施方式或具体例而成,但本发明并不限定于此种实施方式或具体例。此外,本说明书中使用“~”所表示的数值范围意指包含“~”的前后所记载的数值作为下限值及上限值的范围。
以下列举合成例及实施例,更具体地对本发明的特征进行说明。以下所示的材料、处理内容、处理顺序等只要不脱离本发明的主旨便可进行适当变更。因此,本发明的范围不应由以下所示的具体例作限定性地解释。
本发明所提到的简称所代表的化合物如下:
实施例1化合物的合成和发光性能的表征
合成路线1为:
合成路线2:
合成步骤如下:
步骤1——格氏试剂的制备:
在干燥的双口瓶内加入1.336克(0.055摩尔)干燥镁屑、2.326克(0.055摩尔)无水氯化锂,将体系置换为惰性氩气气氛后,加入20毫升超干四氢呋喃溶剂,并滴加0.1毫升的1,2-二溴乙烷。加热至60℃,保持30分钟。逐滴滴加10.000g(0.05摩尔)经干燥的2-溴-1,3,5-三甲基苯,60℃反应两小时,体系变为棕黑色,镁屑消失,得到新制备的1,3,5-三甲基苯基-2-溴化镁格氏试剂溶液。
步骤2——二芳基氯化膦的制备:
将干燥双口瓶内气氛置换为惰性氩气气氛后,加入5.0毫升氯化磷溶液(浓度为2.0摩尔/升的二氯甲烷溶液)和10毫升超干四氢呋喃溶剂,将体系温度降低至-20℃。逐滴滴加10.8毫升按照步骤1新制备的格氏试剂,保持低温0.5小时后自然回升至室温,继续反应3小时,得到二(2,4,6-三甲基苯基)氯化膦溶液。
步骤3——三芳基氯化膦的制备:
对于甲基取代的底物:
在干燥双口瓶内加入1.493克(4.8毫摩尔)5-溴-2-碘-1,3-二甲苯、0.224克(5.28毫摩尔)无水氯化锂,将体系置换为惰性氩气气氛后,加入10毫升超干四氢呋喃溶剂,将体系温度降低至0℃,逐滴滴加2.4毫升正丁基氯化镁溶液(4.8毫摩尔,浓度为2.0摩尔/升的四氢呋喃溶液),0℃反应2小时。保持0℃,逐滴滴加12.4毫升新制备的二-(2,4,6-三甲基苯基)-氯化磷试剂,保持低温0.5小时后自然回升至室温,继续反应3小时。将体系倒入冰水中淬灭,旋蒸除去四氢呋喃后,二氯甲烷作为萃取溶剂萃取。采用碱性氧化铝柱分离,展开剂为石油醚。得到(4-溴-2,6-二甲基苯基)-二(1,3,5-三甲基苯基)膦,为白色粉末,三步综合收率为77%。
对于异丙基取代的底物:
在干燥双口瓶内加入1.439克(4.8毫摩尔)5-溴-2-碘-1,3-二甲苯、0.224克(5.28毫摩尔)无水氯化锂,将体系置换为惰性氩气气氛后,加入10毫升超干四氢呋喃溶剂,将体系温度降低至0℃,逐滴滴加2.4毫升正丁基氯化镁溶液(4.8毫摩尔,浓度为2.0摩尔/升的四氢呋喃溶液),0℃反应2小时。加入0.57克无水氯化亚铜,保持0℃反应1小时。逐滴滴加12.4毫升新制备的二-(2,4,6-三异丙基苯基)-氯化磷试剂,保持低温0.5小时后自然回升至室温,继续反应3小时。将体系倒入冰水中淬灭,旋蒸除去四氢呋喃后,二氯甲烷作为萃取溶剂萃取。采用碱性氧化铝柱分离,展开剂为石油醚。在甲醇中重结晶,得到(4-溴-2,6-二甲基苯基)-二(1,3,5-三异丙基苯基)膦,为白色固体颗粒,三步综合收率为44%。
suzuki偶联
在干燥的三口瓶内加入2克(4.4毫摩尔)(4-溴-2,6-二甲基苯基)-二(1,3,5-三甲基苯基)膦、1.68克(6.6毫摩尔)联频哪醇硼酸酯、1.08克(11.0毫摩尔)无水醋酸钾、0.064克(0.088毫摩尔)[1,1'-双(二苯基膦基)二茂铁]二氯化钯,将体系置换为惰性氩气气氛后加入20毫升无水二氧六环,鼓入氩气除去溶剂中的氧气。加热至110℃反应8小时后,自然冷却至室温。
向三口瓶内继续加入1.3克(4.8毫摩尔)2-氯-4,6-二苯基-1,3,5-三嗪、0.1克(0.086毫摩尔)四(三苯基膦)钯、1.518克无水碳酸钾,补加5毫升去离子水,鼓入氩气除去溶剂中的氧气。加热至110℃反应8小时后,自然冷却至室温。旋蒸除去溶剂,二氯甲烷作为萃取溶剂萃取,采用碱性氧化铝柱分离,展开剂为石油醚:二氯甲烷(50:1)。在甲醇中重结晶,得到对应的suzuki偶联产物。
得到的化合物mp0trz、mp2trz、iprp2trz、mp0phtrz、mp2phtrz、iprp2phtrz的核磁图谱见图1~图6;
其中,图1为化合物mp0trz的h谱和p谱,具体数据如下:1hnmr(400mhz,chloroform-d)δ8.78–8.72(m,4h),8.66(d,j=6.6hz,2h),7.57(qd,j=6.6,4.7hz,8h),6.87(d,j=3.1hz,4h),2.29(s,6h),2.17(s,12h);31pnmr(162mhz,chloroform-d)δ-21.88;
图2为化合物mp2trz的h谱和p谱,具体数据如下:1hnmr(400mhz,chloroform-d)δ8.78–8.74(m,4h),8.33(d,j=3.3hz,2h),7.61–7.53(m,6h),6.83(d,j=3.4hz,4h),2.30–2.27(m,6h),2.25(s,6h),2.11(s,12h),31pnmr(162mhz,chloroform-d)δ-34.20;
图3为化合物iprp2trz的h谱和p谱,具体数据如下:1hnmr(400mhz,chloroform-d)δ8.85–8.71(m,4h),8.38(d,j=3.5hz,2h),7.64–7.53(m,6h),6.96(dt,j=12.7,2.2hz,4h),3.63–3.31(m,4h),2.86(dq,j=13.7,6.9hz,2h),2.39(d,j=1.9hz,6h),1.26–1.14(m,25h),0.66(dd,j=65.4,6.6hz,12h);31pnmr(162mhz,chloroform-d)δ-45.02;
图4为化合物mp0phtrz的h谱和p谱,具体数据如下:1hnmr(400mhz,chloroform-d)δ8.83(d,j=8.2hz,2h),8.79(dd,j=7.9,1.8hz,4h),7.81(d,j=8.2hz,2h),7.65–7.56(m,8h),7.51(s,2h),6.87(d,j=2.9hz,4h),2.29(s,6h),2.18(s,12h);31pnmr(162mhz,chloroform-d)δ-22.68;
图5为化合物mp2phtrz的h谱和p谱,具体数据如下:1hnmr(400mhz,chloroform-d)δ8.85–8.77(m,6h),7.82(d,j=8.5hz,2h),7.60(d,j=7.3hz,6h),7.34(d,j=2.8hz,2h),6.83(d,j=2.6hz,4h),2.28(s,6h),2.16(d,j=30.9hz,18h);31pnmr(162mhz,chloroform-d)δ-35.70;
图6为化合物mp2phtrz的h谱和p谱,具体数据如下:1hnmr(400mhz,chloroform-d)δ8.77–8.68(m,6h),7.75(d,j=8.4hz,2h),7.56–7.47(m,6h),7.26(d,j=3.4hz,2h),6.87(dt,j=17.9,2.3hz,4h),3.42(t,j=12.6hz,4h),2.88–2.68(m,2h),2.25(d,j=1.9hz,6h),1.13(dd,j=20.6,4.8hz,24h),0.60(dd,j=75.2,6.6hz,12h);31pnmr(162mhz,chloroform-d)δ-46.63。
对得到的化合物的光致发光性能进行检测,结果见图7和图8。
图7为实施例1制得的发光分子在甲苯溶液中的光致发光光谱,可以看出基于本发明苯基膦骨架的发光分子可实现从绿光到红光光谱范围内的不同发光波长的发光,可通过调制分子结构调节发光分子的色特性。
图8为实施例1制得的发光分子分布于mcp薄膜中的光致发光光谱,可以看到和溶解于溶液中类似的现象,说明本发明发光在薄膜中也具有优异的光致发光特性。
mcp的分子式为:
图9为本实施例分子的瞬态光致发光光谱衰减曲线,从图中我们看出曲线分为快速部分和慢速部分,是热活性延迟荧光的特性,证明本发明发光分子发射热活性延迟荧光。
实施例2
将实施例1得到的材料制作了有机电致发光元件,该有机电致发光元件采用bepp2作为发光分子的主体材料,并对进行评价。
bepp2的分子式为:
分别得到器件的电压-电流密度-亮度曲线、电致发光光谱和电流密度-外量子效率曲线,结果分别见图10、图11和图12。
图10为得到的器件的电压-电流密度-亮度曲线,可以看出器件能够进行有效的电子空穴注入,器件实现了高亮度的电致发光。
图11为得到的器件的电致发光光谱,可以看出基于本发明分子制备的器件可发射不同波长的光。
图12为器件的电流密度-外量子效率曲线,器件的最大外量子效率均大于5%,也就是荧光器件在20%光取出下利率的情况下的理论值,说明器件实现了电致热活性延迟荧光发射,具有高的发光效率。
器件评价的参数汇总于下表。
- 还没有人留言评论。精彩留言会获得点赞!