一种脱氧氟化试剂的制备方法与流程
1.本发明属于有机合成领域,具体涉及一种脱氧氟化试剂的制备方法。
背景技术:
2.药物化学家研究发现在药物分子中引入氟原子或含氟基团,可以有效改变药物分子的pka值、渗透性、代谢稳定性及脂溶性,对药物分子的体内吸收、分布、代谢以及与生物靶点的相互作用产生影响。得益于许多氟化试剂的发展和具有良好官能团兼容性氟化反应的发现,近几十年来含氟药物研究领域取得了飞速发展。许多临床阶段的新药或者已上市的新药或者上市多年的药物中也含有氟原子。如:艾氟替尼、氟马替尼、罗氟司特、左氧氟沙星、氟维司群等。2018年,美国fda批准了38种小分子药物中,18种含氟药物,占据了其中半壁江山。因此,化学家们开发出越来越多的氟化方法来合成它们。其中,由于天然醇和合成醇的来源都很丰富,脱氧氟化反应成为其中一种非常有吸引力的合成路线。
3.脱氧氟化是指将醇类化合物中的羟基用氟原子进行取代,制备烷基氟化物,其机理一般为醇羟基自身进攻氟化试剂,离去氟负离子,同时o原子转变为离去基的一部分,随后氟负离子对c-o键亲核进攻,实现脱氧氟化历程。
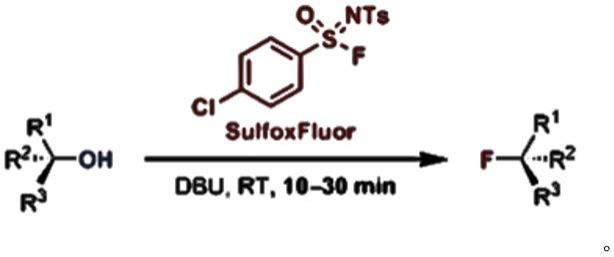
4.n-甲苯磺酰基-4-氯苯磺酰亚胺氟是一种稳定性好,反应性强,有良好的应用前景的脱氧氟化试剂。该脱氧氟化试剂的应用公开在期刊文献european journal 2019(10.1002/chem.201901176)中。反应式如下:
[0005][0006]
上述期刊文献和专利文献cn107245023a中进一步公开了该脱氧氟化试剂的制备方法,以氯苯磺酰氯为原料,经与亚硫酸钠反应、二氯亚砜反应和氯胺t反应制备得到n-甲苯磺酰基-4-氯苯磺酰亚胺氯后,再与氟化钾反应制备得到n-甲苯磺酰基-4-氯苯磺酰亚胺氟。反应式如下:
[0007][0008]
考虑到n-甲苯磺酰基-4-氯苯磺酰亚胺氟的广泛应用前景,本发明开发了一种该脱氧氟化试剂的制备方法。
技术实现要素:
[0009]
本发明提供了一种n-甲苯磺酰基-4-氯苯磺酰亚胺氟的制备方法,本发明是一锅法合成,操作简便,收率高,是一种非常经济且适于工业化生产的制备方法。
[0010]
本发明提供了一种脱氧氟化试剂即n-甲苯磺酰基-4-氯苯磺酰亚胺氟的制备方法。为了实现本发明的技术目的,本发明提供的技术方案为:
[0011]
本发明首先提供了一种n-甲苯磺酰基-4-氯苯磺酰亚胺氯的制备方法。以对氯苯磺酰氯为原料,经一锅法反应制备。反应式为:
[0012][0013]
其中ts为对甲苯磺酰基。
[0014]
本发明上述一锅法反应的过程中,需要加入的其他原料分别为钠盐类、酸、亚砜类、氯胺类等。所述钠盐类可以为碳酸氢钠、亚硫酸钠等;所述酸可以为盐酸;所述亚砜类可以为二氯亚砜;所述氯胺类可以为氯胺t等。反应式如下:
[0015][0016]
较优选地,本发明的一锅法反应过程为:
[0017][0018]
具体为对氯苯磺酰氯经成盐、酸化后,与二氯亚砜和氯胺t反应制备得到n-甲苯磺酰基-4-氯苯磺酰亚胺氯。
[0019]
本发明上述一锅法的反应步骤中,与氯胺t的反应温度为30℃~60℃,较优选地为30℃~35℃。
[0020]
另一方面,本发明提供了n-甲苯磺酰基-4-氯苯磺酰亚胺氟的制备方法。由上述制备得到的n-甲苯磺酰基-4-氯苯磺酰亚胺氯经氟化反应制备得到。反应式为:
[0021][0022]
所述氟化试剂可以为氟化钾、氟化铯、氟化银、氟化铵等。
[0023]
本发明提供的n-甲苯磺酰基-4-氯苯磺酰亚胺氟的制备方法,以对氯苯磺酰氯为原料,经一锅法先制备得到n-甲苯磺酰基-4-氯苯磺酰亚胺氯,后与氟化钾等反应制备得到
n-甲苯磺酰基-4-氯苯磺酰亚胺氟。
[0024]
本发明上述的制备方法,存在如下优势和效果:1.现有技术中的制备方法,存在批量放大反应时,反应时间过长的问题。本发明在批量放大反应时,反应时间大大缩短2.相比于现有技术中的制备方法,溶试剂的用量大大减少,更加经济和适于工业化生产。3.相比于现有技术中的制备方法,本发明制备得到的产品(脱氧氟化试剂)收率大大提高,具备显著地技术效果。4.本发明在氨亚基替磺酰氯即n-甲苯磺酰基-4-氯苯磺酰亚胺氯的制备上,是一锅法的操作步骤,操作简便。5.本发明在氨亚基替磺酰氟即最终产品的制备上,后处理中无需柱层析纯化,加水可以分离产品,收率更高。
具体实施方式
[0025]
为了进一步理解本发明,下面结合实施例对本发明提供的一种脱氧氟化试剂的制备方法进行详细说明。需要理解的是,这些实施例描述只是为进一步详细说明本发明的特征,而不是对本发明范围或本发明权利要求范围的限制。
[0026]
实施例1:
[0027]
往2l四口圆底烧瓶中加入141.12g(2.0eq)碳酸氢钠,105.87g(1.0eq)亚硫酸钠和900ml水,滴加177.6g对氯苯磺酰氯的thf混合液,反应tlc监测,反应完全。静置,加入300ml mtbe(甲基叔丁基醚),萃取分液后取水相,往水相中滴加144ml浓hcl;往反应液中加入1.2l dcm(二氯甲烷),搅拌后萃取分液取有机相,无水硫酸镁干燥,抽滤,直接用于下一步反应。
[0028]
实施例2:
[0029]
将上一步反应液置于3l四口圆底烧瓶,恒压滴液漏斗,n2保护,尾气吸收装置,冰盐浴冷却。滴加134.9g(1.5eq)二氯亚砜;反应完,减压蒸馏浓缩,最终得到461.4g黄绿色溶液,直接用于下一步反应。
[0030]
实施例3:
[0031]
另准备2l四口圆底烧瓶,温度计,氮气保护;往烧瓶中依次加入224.1g(1.17eq)氯胺t,加入1120ml甲苯,添加水浴将內温控制在32~35℃之间,滴加实施例2中制备得到的反应液的甲苯溶液,滴加完毕,滴加过程中,內温维持在30~40℃之间;tlc监测反应完全,室温下静置过夜。抽滤,甲苯淋洗两次;分水、除水;减压蒸馏浓缩,抽滤得到220.84g白色颗粒状固体为n-甲苯磺酰基-4-氯苯磺酰亚胺氯。经计算,一锅法反应总收率为72.3%。
[0032]
实施例4:
[0033]
准备2l三口圆底烧瓶,回流管,温度计,烘干备用;n2保护下,往烧瓶中依次加入220.84g(0.606mol)n-甲苯磺酰基-4-氯苯磺酰亚胺氯,1500ml乙腈,70.5g(1.21mol)kf,3.21g(0.0121mol)18-冠醚-6,45℃油浴恒温搅拌,减压蒸馏浓缩;加入h2o,搅拌、抽滤,淋洗两次得289g类白色晶状固体,置于减压烘箱中过夜,称重得206g,反应收率为97.9%。
[0034]
实施例5:
[0035]
经重复专利cn107245023中的工艺,具体为其实施例i-2中的工艺,具体为n-甲苯磺酰基-4-氯苯磺酰亚胺氯在小试上反应可以制备得到,当在批量放大反应时,存在反应时间长的问题。本发明很好的解决了批量放大时反应时间长的问题,具备显著地技术效果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1