一种化合物、其制备方法及应用以及抗类风湿性关节炎的药物与流程
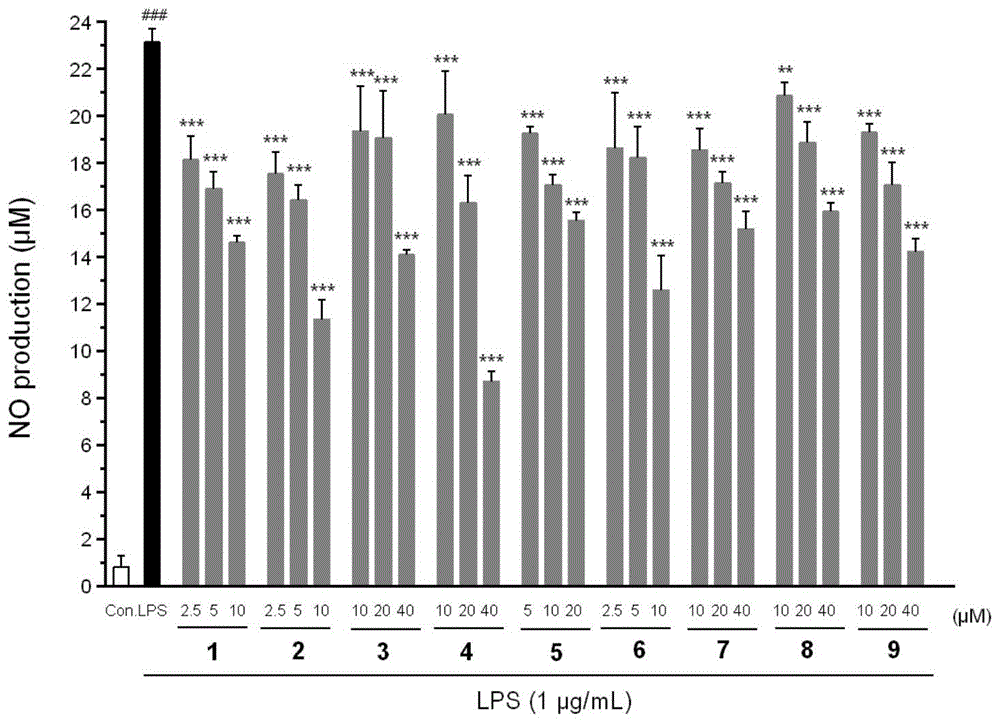
本申请为分案申请,母案的申请号为201910569463.2,名称为“一种化合物、其制备方法及应用以及抗类风湿性关节炎的药物”。本发明涉及天然药物及医药
技术领域:
,且特别涉及一种化合物、其制备方法及应用以及抗类风湿性关节炎的药物。
背景技术:
:类风湿性关节炎(rheumatoidarthritis,ra)是一种以慢性炎症应答和关节破坏为主的多系统自身免疫性疾病。其主要病理特征为慢性持续性的滑膜炎症,滑膜细胞增生,炎症细胞浸润,血管翳形成,软骨和骨组织的侵蚀和破坏,最终导致关节组织结构破坏、畸形和功能丧失,造成不同程度的残疾,严重影响患者生活质量。ra病变不仅侵蚀骨关节,还累及各脏器,伴随关节外的多系统病变。ra的全球发病率在1%-2%之间,在我国,发病率约为0.2%-0.37%,是造成我国人群致残和丧失劳动力的主要疾病之一。目前,类风湿性关节炎主要以非甾体类抗炎药、改善病情抗风湿药、糖皮质激素等药物治疗为主,但这些药物疗效有限,毒副作用明显,以至于不能长期服用,因此,寻找与研发新型高效低毒的药物用于预防和治疗类风湿性关节炎的炎症发展及关节损伤仍然迫在眉睫。天然产物具有来源广泛、成分丰富及不良反应少等特点,越来越受到国内外研究学者的关注。目前从天然产物中尝试寻找抗类风湿性关节炎药物已经成为研究热点。烟豆(glycinetabacina(labill.)benth)是豆科(leguminosae)大豆属(glycine)多年生草本植物,又称澎湖大豆、澎湖一条根、尖叶一条根、长叶银豆等,分布于澳大利亚、南洋诸岛、及中国东南沿海和台湾地区等。烟豆具有祛风湿、壮筋骨、益脾肾之功效,民间用以治疗风湿骨痛、腰膝酸软诸症。但目前对烟豆的化学成分和药理作用的研究十分缺乏,烟豆的药用价值未得到足够重视。因此,开展烟豆的药理和药效物质基础研究,尤其是新化合物的开发,对烟豆药用资源的挖掘与利用具有重要意义。技术实现要素:本发明的目的在于提供一种化合物,旨在提供具有预防和治疗类风湿性关节炎功效的新化合物。本发明的另一目的在于提供一种化合物的制备方法,旨在从烟豆中提取具有预防和治疗类风湿性关节炎功效的活性成分。本发明的另三目的在于提供上述化合物的用途以及由上述化合物作为活性成分的抗类风湿性关节炎的药物。本发明解决其技术问题是采用以下技术方案来实现的。本发明提出了一种化合物,其结构式为:其中,r1表示甲氧基或羟基;r2表示r3表示或h;r4表示r5表示甲氧基或羟基。本发明还提出一种化合物的制备方法,包括如下步骤:从烟豆中提取上述化合物。本发明还提出上述化合物在制备预防和治疗炎症疾病的药物中的应用;优选地,炎症疾病为类风湿性关节炎。本发明还提出一种抗类风湿性关节炎的药物,其活性成分包括上述化合物。本发明实施例提供一种化合物的有益效果是:发明人通过不断探究发现具有抗类风湿性关节炎功效的新化合物,能够有效抑制lps刺激的一氧化氮的产生,具有较强的体外抗炎活性,同时具有很好的缓解类风湿性关节炎滑膜炎症的能力。本发明还提供了一种化合物的制备方法,其通过从烟豆中提取上述化合物,从烟豆复杂的成分组成中提取活性成分,对烟豆药用资源的挖掘与利用具有重要意义。本发明还提供了上述化合物的用途以及以上述化合物作为活性成分的药物,具有很好的市场应用价值。附图说明为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。图1为在raw264.7细胞中,安全浓度范围内的化合物1-9对lps刺激产生的一氧化氮水平的影响;图2为在sw982细胞中,安全浓度范围内的化合物1-9对il-1β刺激产生的tnf-α水平的影响;图3为安全浓度范围内的化合物1、2、7、8、9对rankl刺激的raw264.7细胞向破骨细胞分化过程的影响;图4为第一化合物的1h-nmr谱;图5为第一化合物的13c-nmr谱;图6为第一化合物的hsqc谱;图7为第一化合物的hmbc谱;图8为第二化合物的1h-nmr谱;图9为第二化合物的13c-nmr谱;图10为第二化合物的hsqc谱;图11为第二化合物的hmbc谱;图12为第三化合物的1h-nmr谱;图13为第三化合物的13c-nmr谱;图14为第三化合物的hsqc谱;图15为第三化合物的hmbc谱;图16为第四化合物的1h-nmr谱;图17为第四化合物的13c-nmr谱;图18为第四化合物的hsqc谱;图19为第四化合物的hmbc谱;图20为第五化合物的1h-nmr谱;图21为第五化合物的13c-nmr谱;图22为第五化合物的hsqc谱;图23为第五化合物的hmbc谱;图24为第六化合物的1h-nmr谱;图25为第六化合物的13c-nmr谱;图26为第六化合物的hsqc谱;图27为第六化合物的hmbc谱;图28为第七化合物的1h-nmr谱;图29为第七化合物的13c-nmr谱;图30为第七化合物的hsqc谱;图31为第七化合物的hmbc谱;图32为第八化合物的1h-nmr谱;图33为第八化合物的13c-nmr谱;图34为第八化合物的hsqc谱;图35为第八化合物的hmbc谱;图36为第九化合物的1h-nmr谱;图37为第九化合物的13c-nmr谱;图38为第九化合物的hsqc谱;图39为第九化合物的hmbc谱。具体实施方式为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。下面对本发明实施例提供的一种化合物、其制备方法及应用以及抗类风湿性关节炎的药物进行具体说明。本发明实施例提供的一种化合物,其结构式为:其中,r1表示甲氧基或羟基;r2表示r3表示或h;r4表示r5表示甲氧基或羟基。需要说明的是,以上化合物为新化合物,能够显著缓解脂多糖和白细胞介素-1β刺激的细胞炎症反应,并且具有抑制破骨细胞分化的能力,显示该类化合物在制备抗类风湿性关节炎药物中的应用前景。发明人对符合上述结构的多种化合物进行了研究,筛选出效果最为突出的9种化合物,可以概括为以下结构:具体地,该化合物其选自第一化合物、第二化合物、第三化合物、第四化合物、第五化合物、第六化合物、第七化合物、第八化合物、第九化合物中的任意一种;其中,第一化合物至第九化合物的结构式依次为:以上第一化合物至第九化合物均来自烟豆中,其为烟豆的活性成分,使烟豆具有抗类风湿关节炎的效果。本发明实施例还提供了一种化合物的制备方法,包括:从烟豆中提取上述化合物;优选地,提取的化合物包括第一化合物、第二化合物、第三化合物、第四化合物、第五化合物、第六化合物、第七化合物、第八化合物和第九化合物。具体包括如下步骤:s1、浸膏制备将烟豆粉碎后进行醇提再浓缩得醇提浸膏。为了使活性成分更多的提取出来,利用体积分数为90-98%的乙醇溶剂进行至少3次萃取至提取液颜色较浅,合并萃取液,然后过滤并进行减压浓缩即可。优选地,提取时的料液比为1:5-10,料液比控制在上述范围内能够使烟豆中的有效成分更充分的提取出来,提高活性成分的提取率。具体地,提取方法为冷浸提取、加热回流提取或超声辅助提取等现有的提取方法。s2、溶剂萃取将醇提浸膏与水混合后依次采用石油醚和乙酸乙酯进行萃取得石油醚萃取部分和乙酸乙酯萃取部分。发明人发现,采用此种萃取顺序能够将活性成分更多地集中在乙酸乙酯萃取部分中,便于后续进行分离纯化。在一些实施例中,溶剂萃取过程中是分别用石油醚、乙酸乙酯、水饱和正丁醇进行萃取,得到石油醚萃取部分、乙酸乙酯萃取部分、正丁醇萃取部分和水萃取部分。通过多次萃取能够将烟豆中的成分进行更有效的分离,提升最终提取的活性成分的纯度,其中萃取剂的选择以及萃取顺序的设定均以上述方案为宜,在此条件下能够有效提高提取产品的纯度。s3、层析分段将乙酸乙酯萃取部分进行梯度洗脱得到11个洗脱部分,活性成分集中于这11个洗脱部分,洗脱部分的排序可以是自上而下或自左至右依次编号。优选地,层析分段过程中是利用硅胶色谱柱进行分段,用石油醚-乙酸乙酯系统进行梯度洗脱,发明人发现采用石油醚-乙酸乙酯系统能够使得到的洗脱部分在后续的分离提纯时更加方便,各化合物的提取率和纯度均较为理想。优选地,用石油醚-乙酸乙酯系统进行梯度洗脱时,石油醚:乙酸乙酯的洗脱梯度为10:1、5:1、3:1、2:1、1:1、1:2和1:5。发明人对梯度洗脱时的洗脱体系配比进行了优化,采用上述的洗脱梯度能够使洗脱部分更好的分层。s4、纯化分离利用第一洗脱部分、第二洗脱部分和第三洗脱部分合并纯化得第三化合物;利用第五洗脱部分纯化得第一化合物;利用第六洗脱部分纯化得第四化合物;利用第七洗脱部分纯化得第六化合物、第七化合物和第二化合物;利用第九洗脱部分纯化得第五化合物、第八化合物和第九化合物。发明人经过大量的实验探究,优化了纯化步骤,采用上述的纯化过程能够更大程度上提升化合物的提取率和纯度,减少原料浪费。为了进一步提升9种化合物的提取率和纯度,发明人对各个洗脱部分的分离提纯步骤进行了优化。其中,利用第一洗脱部分、第二洗脱部分和第三洗脱部分合并纯化得第三化合物的过程包括:将三个洗脱部分合并后上柱层析洗脱,然后利用制备薄层色谱纯化。优选地,三个洗脱部分对应的洗脱液为二氯甲烷和甲醇的混合液;更优选地,二氯甲烷和甲醇的体积比为1:0.5-1.5。采用二氯甲烷和甲醇的混合液为洗脱液产品的收率更高,发明人通过调控洗脱液的配比进一步第三化合物的收率。具体地,多次制备薄层色谱的色谱条件中,第一次制备的条件为:石油醚和乙酸乙酯的体积比为4-6:1,第二次制备的条件为:石油醚和二氯甲烷的体积比为1-3:3;三个洗脱部分对应的柱层析为sephadexlh-20柱层析。制备薄层色谱的色谱条件对第三化合物的收率和纯度均有一定影响,将色谱条件控制在上述范围内能够更高效地分离得到第三化合物,收率大致为0.0003‰。其中,利用第五洗脱部分纯化得第一化合物的过程中包括:将第五洗脱部分浓缩5-8天,过滤结晶。经过长时间的浓缩有大量的晶体生成,将此部分晶体收集过滤得到纯净的第一化合物,收率大致为0.015‰。其中,利用第六洗脱部分纯化得第四化合物的过程包括:将第六洗脱部分上柱层析,洗脱液经tlc检识后合并得五种流份,取第四种流份利用制备薄层色谱纯化;优选地,第四种流份对应的制备薄层色谱的色谱条件:石油醚和丙酮的体积比为2-4:1;优选地,第六洗脱部分对应的柱层析为sephadexlh-20柱层析。第四化合物的分离需要经过多步分离,方可达到更高纯度,以上分离步骤是发明人经过反复实践得出,通过选择特定的流份并优化色谱条件使第四化合物的收率和纯度达到最佳,收率大致为0.0005‰。其中,利用第七洗脱部分纯化得第六化合物、第七化合物和第二化合物的过程包括:将第七洗脱部分上sephadexlh-20柱层析,洗脱液经tlc检识后合并得五种流份7a-7e,取流份7a上toyopealhw-40柱层析,洗脱液经tlc检识后合并得五种流份7a1-7a5,取流份7a3和7a4分别经制备液相色谱纯化后获得第六化合物和第七化合物(收率分别为0.0005‰和0.0009‰),取流份7c再次上sephadexlh-20柱层析后得第二化合物(收率大致为0.027‰)。第六化合物、第七化合物和第二化合物均从第七洗脱部分进行提纯,提纯步骤较为繁琐,为达到更高的收率发明人进行了不断探究,以上的提纯分离步骤为优化后的步骤。发明人进一步优化了液相色谱制备条件以进一步提升分离效果,在获得第六化合物和第七化合物过程中液相色谱的制备条件为:采用18c-反向色谱柱,流速为7-9ml/min;优选地,第六化合物对应的洗脱液为甲醇和水的混合物,且甲醇和水的体积比为90:8-12;优选地,第七化合物对应的洗脱液为甲醇和水的混合物,且甲醇和水的体积比为95:4-6。其中,利用第九洗脱部分纯化得第五化合物、第八化合物和第九化合物的过程包括:将第九洗脱部分进行硅胶色谱柱层析,并用石油醚-丙酮系统进行梯度洗脱得七种洗脱流份9a-9g;取流份9d上sephadexlh-20柱层析,洗脱液经tlc检识后合并得三种流份9d1-9d3;取流份9d2经制备液相色谱分离得第五化合物(收率大致为0.0007‰);取流份9e上sephadexlh-20柱层析,洗脱液经tlc检识后合并得三种流份9e1-9e3;取流份9e3上toyopealhw-40柱层析得三种流份9e3a-9e3c,取流份9e3b经制备液相色谱纯化后获得第八化合物和第九化合物(收率分别为0.0004‰和0.0007‰)。第五化合物、第八化合物和第九化合物均提纯自第九洗脱部分,此部位提纯工艺更为繁琐,发明人同样是经过反复研究优化工艺步骤,提升了第五化合物、第八化合物和第九化合物收率和纯度。优选地,用石油醚-丙酮系统进行梯度洗脱时,石油醚:丙酮的洗脱梯度为10:1、6:1、4:1、3:1、2:1、1:1、2:3、1:2。采用以上的洗脱梯度能够使第九洗脱部分更好地分离,提升三种化合物的产率。发明人进一步优化了液相色谱制备条件以进一步提升分离效果,在获得第五化合物、第八化合物和第九化合物过程中液相色谱的制备条件为:采用18c-反向色谱柱,流速为7-9ml/min;更优选地,第五化合物对应的洗脱液为甲醇和水的混合物,且甲醇和水的体积比为90:8-12;更优选地,第八化合物对应的洗脱液为甲醇和水的混合物,且甲醇和水的体积比为70:25-35;更优选地,第九化合物对应的洗脱液为甲醇和水的混合物,且甲醇和水的体积比为70:25-35。需要说明的是,以上sephadexlh-20柱层析的洗脱剂为二氯甲烷和甲醇的混合液,优选地,二氯甲烷和甲醇的体积比为1:0.5-1.5;以上toyopealhw-40柱层析的洗脱剂为甲醇。按照以上优化后的步骤与条件对第一至第九化合物进行分离纯化,得到的化合物纯度均在90%以上。s5、表征在纯化分离后对提取的化合物进行核磁谱图和质谱表征以鉴定化合物结构;更优选地,核磁谱图选自1h-nmr、13c-nmr、hmbc、hsqc或noesy;更优选地,质谱选自hr-esi-ms或esi-ms。在分离提纯完成后可以进行表征,以确定提取的化合物组成。本发明实施例还提供了上述化合物在制备预防和治疗炎症疾病的药物中的应用;优选地,炎症疾病为类风湿性关节炎。测试9种coumestan类新化合物的体外抗炎和抗类风湿性关节炎活性,药效实验表明,在脂多糖(lps)刺激的raw264.7细胞(小鼠单核巨噬细胞白血病细胞)炎症模型中,在化合物的安全浓度范围内,9种化合物能够不同程度地抑制lps刺激的一氧化氮的产生,且具有量效关系,表明9种化合物具有较强的体外抗炎活性;在白细胞介素-1β(il-1β)刺激的sw982细胞(人滑膜肉瘤细胞)炎症模型中,在各化合物的安全浓度范围(5-20μm)内,9种化合物能够不同程度地抑制il-1β刺激的促炎因子tnf-α的产生,表明了9种化合物具有缓解类风湿性关节炎滑膜炎症的能力;同时建立核因子κb受体活化因子配体(rankl)刺激的raw264.7细胞向破骨细胞分化模型,发现在各化合物的安全浓度(5-20μm)范围内,与rankl模型组相比,第一化合物、第二化合物、第七化合物、第八化合物和第九化合物能够不同程度地降低抗酒石酸酸性磷酸酶(trap)活性,抑制了raw264.7细胞向破骨细胞的分化过程。本发明实施例还提供了一种抗类风湿性关节炎的药物,其活性成分包括上述化合物;可以将上述9种化合物作为药物的活性成分制成抗类风湿性关节炎的药物,药物的制剂方式为丸剂、颗粒剂、片剂、囊剂或注射剂。以下结合实施例对本发明的特征和性能作进一步的详细描述。仪器与材料:烟豆药材购于福建省福清市,通过dna条形码鉴定植物种属(中国医学科学院药用植物研究所韩建萍博士实验室完成);柱色谱用硅胶(200-300目);薄层色谱用硅胶gf254(10-40μm)(青岛海洋化工有限公司);柱色谱用sephadexlh-20填料;柱色谱用toyopealhw-40填料;制备色谱柱cosmosil5c18-ms-ii20id×250mm;制备液相色谱shimadzupreparativesystemlc-20ap;核磁共振仪:bruker,600mhz;高分辨质谱thermoltqorbitrapxlhybridftms。实施例1本实施例提供一种化合物的制备方法,其包括以下步骤:(1)乙醇浸膏提取:将5kg烟豆全草干燥后粉碎,装入提取桶,按料液比1:5的95%乙醇冷浸提取3次,每次3天,至提取液颜色较浅,合并滤液,过滤,减压浓缩得乙醇浸膏184g。(2)溶剂萃取:将烟豆乙醇浸膏用800ml蒸馏水制备水混悬液,分别经石油醚、乙酸乙酯和水饱和正丁醇萃取,得石油醚萃取部分29.0g、乙酸乙酯萃取部分22.8g、正丁醇萃取部分30.1g和水萃取部分74.3g。(3)硅胶柱层析分段:将烟豆乙酸乙酯萃取部分上200-300目硅胶色谱柱(70×245mm)进行分段,用石油醚-乙酸乙酯系统(10:1,5:1,3:1,2:1,1:1,1:2,1:5,乙酸乙酯)进行梯度洗脱,得到11个洗脱部分fr.1(0.25g),fr.2(0.14g),fr.3(0.15g),fr.4(0.51g),fr.5(0.30g),fr.6(0.51g),fr.7(1.06g),fr.8(4.09g),fr.9(4.19g),fr.10(3.73g),fr.11(5.73g)。(4)将洗脱部分fr.5浓缩放置7天,待大量结晶生成后过滤结晶得第一化合物(72mg);将洗脱部分fr.1、fr.2和fr.3合并后上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1洗脱,洗脱液经tlc检识后合并,上多次制备薄层制备色谱(石油醚:乙酸乙酯=5:1;石油醚:二氯甲烷=2:3),纯化获得第三化合物(1.4mg);将组分fr.6上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1洗脱,洗脱液经tlc检识后合并得流份6a-6e,流份6d上多次制备薄层制备色谱(石油醚:丙酮=3:1)纯化后获得第四化合物(2.3mg);将组分fr.7上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1洗脱,洗脱液经tlc检识后合并得流份7a-7e,流份7a上toyopealhw-40柱层析,甲醇洗脱,洗脱液经tlc检识后合并得流份7a1-7a5,流份7a3和7a4经制备液相色谱纯化后获得第六化合物(2.6mg,甲醇:水=90:10)和第七化合物(4.5mg,甲醇:水=95:5),流份7c再次上sephadexlh-20柱层析,二氯甲烷;甲醇=1:1洗脱,得第二化合物(135mg)。(5)将组分fr.9进行硅胶色谱柱层析(36×210mm)继续分段,用石油醚-丙酮系统(10:1,6:1,4:1,3:1,2:1,1:1,2:3,1:2)进行梯度洗脱,得到7个洗脱流份9a-9g。流份9d上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1洗脱,洗脱液经tlc检识后合并得流份9d1-9d3,流份9d2经制备液相色谱分离得第五化合物(3.2mg,甲醇:水=90:10),流份9e上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1洗脱,得流份9e1-9e3,流份9e3进一步上toyopealhw-40柱层析,甲醇洗脱,得流份9e3a-9e3c,流份9e3b经制备液相色谱纯化后获得第八化合物(1.9mg,甲醇:水=70:30)和第九化合物(3.4mg,甲醇:水=70:30)。实施例2本实施例提供一种化合物的制备方法,其包括以下步骤:(1)乙醇浸膏提取:将5kg烟豆全草干燥后粉碎,装入提取桶,按料液比1:10的90%乙醇冷浸提取3次,每次3天,至提取液颜色较浅,合并滤液,过滤,减压浓缩得乙醇浸膏。(2)溶剂萃取:将烟豆乙醇浸膏用800ml蒸馏水制备水混悬液,分别经石油醚、乙酸乙酯和水饱和正丁醇萃取,得石油醚萃取部分、乙酸乙酯萃取部分、正丁醇萃取部分和水萃取部分。(3)硅胶柱层析分段:将烟豆乙酸乙酯萃取部分上200-300目硅胶色谱柱(70×245mm)进行分段,用石油醚-乙酸乙酯系统(10:1,5:1,3:1,2:1,1:1,1:2,1:5,乙酸乙酯)进行梯度洗脱,得到11个洗脱部分fr.1,fr.2,fr.3,fr.4,fr.5,fr.6,fr.7,fr.8,fr.9,fr.10,fr.11。(4)将洗脱部分fr.5浓缩放置5天,待大量结晶生成后过滤结晶得第一化合物;将洗脱部分fr.1、fr.2和fr.3合并后上sephadexlh-20柱层析,二氯甲烷:甲醇=1:0.5洗脱,洗脱液经tlc检识后合并,上多次制备薄层制备色谱(石油醚:乙酸乙酯=4:1;石油醚:二氯甲烷=1:3),纯化获得第三化合物;将组分fr.6上sephadexlh-20柱层析,二氯甲烷:甲醇=1:0.5洗脱,洗脱液经tlc检识后合并得流份6a-6e,流份6d上多次制备薄层制备色谱(石油醚:丙酮=2:1)纯化后获得第四化合物;将组分fr.7上sephadexlh-20柱层析,二氯甲烷:甲醇=1:0.5洗脱,洗脱液经tlc检识后合并得流份7a-7e,流份7a上toyopealhw-40柱层析,甲醇洗脱,洗脱液经tlc检识后合并得流份7a1-7a5,流份7a3和7a4经制备液相色谱纯化后获得第六化合物(甲醇:水=90:8)和第七化合物(甲醇:水=95:4),流份7c再次上sephadexlh-20柱层析,二氯甲烷;甲醇=1:0.5洗脱,得第二化合物。(5)将组分fr.9进行硅胶色谱柱层析(36×210mm)继续分段,用石油醚-丙酮系统(10:1,6:1,4:1,3:1,2:1,1:1,2:3,1:2)进行梯度洗脱,得到7个洗脱流份9a-9g。流份9d上sephadexlh-20柱层析,二氯甲烷:甲醇=1:0.5洗脱,洗脱液经tlc检识后合并得流份9d1-9d3,流份9d2经制备液相色谱分离得第五化合物(甲醇:水=90:8),流份9e上sephadexlh-20柱层析,二氯甲烷:甲醇=1:0.5洗脱,得流份9e1-9e3,流份9e3进一步上toyopealhw-40柱层析,甲醇洗脱,得流份9e3a-9e3c,流份9e3b经制备液相色谱纯化后获得第八化合物(甲醇:水=70:25)和第九化合物(甲醇:水=70:25)。实施例3本实施例提供一种化合物的制备方法,其包括以下步骤:(1)乙醇浸膏提取:将5kg烟豆全草干燥后粉碎,装入提取桶,按料液比1:8的98%乙醇冷浸提取3次,每次3天,至提取液颜色较浅,合并滤液,过滤,减压浓缩得乙醇浸膏。(2)溶剂萃取:将烟豆乙醇浸膏用800ml蒸馏水制备水混悬液,分别经石油醚、乙酸乙酯和水饱和正丁醇萃取,得石油醚萃取部分、乙酸乙酯萃取部分、正丁醇萃取部分和水萃取部分。(3)硅胶柱层析分段:将烟豆乙酸乙酯萃取部分上200-300目硅胶色谱柱(70×245mm)进行分段,用石油醚-乙酸乙酯系统(10:1,5:1,3:1,2:1,1:1,1:2,1:5,乙酸乙酯)进行梯度洗脱,得到11个洗脱部分fr.1,fr.2,fr.3,fr.4,fr.5,fr.6,fr.7,fr.8,fr.9,fr.10,fr.11。(4)将洗脱部分fr.5浓缩放置8天,待大量结晶生成后过滤结晶得第一化合物;将洗脱部分fr.1、fr.2和fr.3合并后上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1.5洗脱,洗脱液经tlc检识后合并,上多次制备薄层制备色谱(石油醚:乙酸乙酯=6:1;石油醚:二氯甲烷=3:3),纯化获得第三化合物;将组分fr.6上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1.5洗脱,洗脱液经tlc检识后合并得流份6a-6e,流份6d上多次制备薄层制备色谱(石油醚:丙酮=4:1)纯化后获得第四化合物;将组分fr.7上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1.5洗脱,洗脱液经tlc检识后合并得流份7a-7e,流份7a上toyopealhw-40柱层析,甲醇洗脱,洗脱液经tlc检识后合并得流份7a1-7a5,流份7a3和7a4经制备液相色谱纯化后获得第六化合物(甲醇:水=90:12)和第七化合物(甲醇:水=95:6),流份7c再次上sephadexlh-20柱层析,二氯甲烷;甲醇=1:1.5洗脱,得第二化合物。(5)将组分fr.9进行硅胶色谱柱层析(36×210mm)继续分段,用石油醚-丙酮系统(10:1,6:1,4:1,3:1,2:1,1:1,2:3,1:2)进行梯度洗脱,得到7个洗脱流份9a-9g。流份9d上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1.5洗脱,洗脱液经tlc检识后合并得流份9d1-9d3,流份9d2经制备液相色谱分离得第五化合物(甲醇:水=90:12),流份9e上sephadexlh-20柱层析,二氯甲烷:甲醇=1:1.5洗脱,得流份9e1-9e3,流份9e3进一步上toyopealhw-40柱层析,甲醇洗脱,得流份9e3a-9e3c,流份9e3b经制备液相色谱纯化后获得第八化合物(甲醇:水=70:35)和第九化合物(甲醇:水=70:35)。试验例1coumestan类新化合物对lps刺激的raw264.7细胞一氧化氮生成的影响实验。主要实验材料:上述实施例1中提取分离得到的9个coumestan类新化合物;raw264.7细胞购自中国科学院细胞库;一氧化氮测定试剂盒购自碧云天生物技术公司;胎牛血清(fbs)和dmem培养基均购自gibco公司。实验方法:实验设control组,lps组,各药物实验组。取长至70%-80%的raw264.7细胞,细胞刮刮下,吹成单细胞悬液,加入含10%fbs的dmem培养基调整细胞浓度为1×105个/ml,接种于96孔板中,每孔100μl,置于37℃、5%co2培养箱内培养过夜。待细胞贴壁后,吸弃上清液,control组加入100μldmem培养基,lps模型组加入100μl含1μg/mllps的dmem培养基,各药物实验组加入100μl含各浓度化合物和1μg/mllps的dmem培养基。37℃,5%co2孵育24小时。培养结束后取上清100μl转至干净96孔板中,分别加入griess试剂a液和b液各50μl,避光显色5min,540nm下用酶标仪测各孔吸光度(od)值,重复3次实验。实验结果:实验结果见图1。各coumestan类新化合物在图1所示的浓度内对raw264.7细胞无细胞毒性。与control组相比,raw264.7细胞经lps刺激后,细胞上清液中的一氧化氮水平明显升高,证明造模非常成功。但在药物实验组中,在各化合物的安全浓度范围内,各coumestan类新化合物处理24小时后均不同程度地降低了一氧化氮产生,且呈现量效关系,表明第一化合物-第九化合物均具有较强的体外抗炎活性。试验例2coumestan类新化合物对il-1β刺激的sw982细胞tnf-α生成的影响实验。主要实验材料:上述实施例1中提取分离得到的9个coumestan类新化合物;sw982细胞购自中国科学院细胞库;tnf-αelisa试剂盒购自biolegend公司;胎牛血清(fbs)和dmem培养基均购自gibco公司。实验方法:实验设control组,il-1β模型组,各药物实验组。取长至70%-80%的sw982细胞,胰酶消化,吹成单细胞悬液,加入含10%fbs的dmem培养基调整细胞浓度为2×105个/ml,接种于96孔板中,每孔100μl,37℃、5%co2培养过夜。待细胞贴壁后,吸弃上清液,空白组加入100μldmem培养基,il-1β模型组加入100μl含10ng/mlil-1β的dmem培养基,各药物实验组加入100μl含各浓度化合物+10ng/mlil-1β的dmem培养基。37℃,5%co2孵育18小时。培养结束后取上清100μl转至干净96孔板中,按如下提出的elisa测定试剂盒步骤测定细胞上清液中tnf-α水平,重复3次实验。a.新鲜配制包被抗体,分别加入100μl包被抗体至各elisa试剂盒检测孔内,4度封闭孵育16-18h。b.吸弃包被抗体,每孔加入300μl洗涤缓冲液进行清洗,重复4次。200μl1×assaydiluent室温孵育1小时。c.吸弃assaydiluent,每孔加入300μl洗涤缓冲液进行清洗,重复4次。吸取100μl细胞上清液或标准品至各孔,室温封闭孵育2小时。d.吸弃细胞上清液,每孔加入300μl洗涤缓冲液进行清洗,重复4次。每孔加入100μltnf-αdetectionantibodysolution,室温封闭孵育1小时。e.吸弃tnf-αdetectionantibody,每孔加入300μl洗涤缓冲液进行清洗,重复4次。每孔加入100μlavidin-hrpsolution,室温封闭孵育30分钟。f.吸弃tnf-αavidin-hrpsolution,每孔加入300μl洗涤缓冲液进行清洗,重复5次。每孔加入100μl新鲜配制的tmbsubstratesolution,室温避光孵育15分钟。g.每孔加入100μl反应终止液,轻轻摇动检测板混匀,迅速于酶标光度计中检测450nm波长处的吸光光度值(od值),根据所得读数绘制标准曲线进行计算。实验结果:实验结果见图2。各coumestan类新化合物在图2所示的浓度内对sw982细胞无细胞毒性。与control组相比,sw982细胞经il-1β刺激后,细胞上清液中的tnf-α水平明显升高,证明造模非常成功。但在各药物实验组中,在化合物的安全浓度范围内,经coumestan类新化合物处理18小时后,各组细胞上清液中的tnf-α水平不同程度地降低,其中化合物1、2、9(即第一化合物、第二化合物和第九化合物)显著抑制tnf-α的产生,化合物3、5、6和8的活性次之,化合物4和7的活性较弱。结果表明,烟豆中coumestan类化合物具有显著抑制类风湿性关节炎的滑膜炎症的能力。试验例3coumestan类新化合物对rankl刺激的raw264.7细胞向破骨细胞分化的影响实验。主要实验材料:上述实施例1中提取分离得到的九9个coumestan类新化合物;raw264.7细胞购自中国科学院细胞库;抗酒石酸酸性磷酸酶(trap)检测试剂盒购自碧云天生物技术公司;胎牛血清(fbs)和dmem培养基均购自gibco公司。实验方法:实验设control组,rankl模型组,药物实验组。取长至70%-80%的raw264.7细胞,细胞刮刮下,吹成单细胞悬液,加入含10%fbs的dmem培养基调整细胞浓度为2×104个/ml,接种于96孔板中,每孔100μl,37℃、5%co2培养过夜。待细胞贴壁后,吸去上清液,空白组加入100μldmem培养基,rankl模型组加入100μl含50ng/mlrankl的dmem培养基,药物实验组加入100μl含各浓度化合物+50ng/mlrankl的dmem培养基。每2天更换一次诱导液,培养4-5天。诱导结束后,吸弃上清,pbs漂洗两次,加入细胞裂解液(无抑制剂)裂解细胞,按如下提出的抗酒石酸酸性磷酸酶(trap)检测试剂盒步骤测定细胞内trap活性,重复3次实验。a.样品准备及稀释:采用细胞裂解液(无抑制剂)裂解细胞,如果样品中含有较高活性的trap,可以使用原有的裂解液或pbs等进行稀释,备用。b.参考下表使用96孔板设置空白对照孔、标准品孔和样品孔。标准品的用量分别为4、8、16、24、32和40微升,样品通常可以直接加40μl。表1空白对照孔、标准品孔和样品孔的设置标准空白对照标准品样品检测缓冲液40μl(80-x)μl(40-y)μl显色底物40μl-40μl酒石酸溶液5μl5μl5μl样品--yμl标准品工作液-xμl-c.用枪头轻轻吹打混匀,也可借助摇床进行混匀。d.37℃孵育20分钟。e.每孔加入160μl反应终止液终止反应。此时,标准品或有酒石酸酸性磷酸酶活性的孔会呈现不同深浅的黄色。f.在405nm测定吸光度。g.酸性磷酸酶活性单位的定义:在ph4.8,37℃条件下,每分钟水解para-nitrophenylphosphate显色底物产生1微摩尔p-nitrophenol所需的酸性磷酸酶的量定义为一个酶活力单位。h.根据酶活性定义,计算出样品中的抗酒石酸酸性磷酸酯酶活性。实验结果:抗酒石酸酸性磷酸酶是一种糖基化的含金属蛋白酶,在破骨细胞(osteoclast)和破软骨细胞(chondroclast)中高表达。抗酒石酸酸性磷酸酶可以被破骨细胞释放到血液中,几乎被认为是机体破骨活性的唯一血液指标。因此,检测细胞内trap的活性可以体现coumestan类新化合物抑制破骨细胞分化的能力。实验结果见图3。各coumestan类新化合物在图3所示的浓度内对raw264.7细胞无细胞毒性。与control组相比,raw264.7细胞经rankl刺激后,细胞内的trap的活性明显升高,证明成功建立了raw264.7细胞向破骨细胞分化模型。但在各给药组中,在化合物的安全浓度范围内,经coumestan类新化合物1-2和化合物7-9共孵育后,各给药组细胞内的trap活性明显降低,且以化合物2和9的作用最为显著。结果表明,烟豆中coumestan类化合物具有显著抑制破骨细胞分化的能力。以下通过药效实验证明本发明的有益效果。实验例1~3实验结果用mean±sd表示。#p<0.05,##p<0.01,###p<0.01,与对照组相比;*p<0.05,**p<0.01,***p<0.001与模型组相比。p<0.05为差异有统计学意义。试验例4对实施例1中制备得到的9种化合物进行表征,测试第一化合物-第九化合物的理化性质,多种核磁谱图(1h-nmr,13c-nmr,hmbc,hsqc,noesy)和hr-esi-ms解析结构。化合物的理化性质和谱图数据如下:第一化合物:白色针晶;hr-esi-msm/z4417.16876[m-h]-(calcd.forc26h25o5,417.16965);nmr(600mhz,dmso-d6)数据见表2;1h-nmr谱(600mhz,dmso-d6)、13c-nmr谱(150mhz,dmso-d6);hsqc谱(600mhz,dmso-d6)以及hmbc谱(600mhz,dmso-d6)如图4-7所示,未见文献报道,为新化合物,命名为glytacoumestana。第二化合物:浅色固体;hr-esi-msm/z403.15323[m-h]-(calcdforc25h23o5,403.15400);nmr(600mhz,dmso-d6)数据见表2;1h-nmr谱(600mhz,cd3od)、13c-nmr谱(150mhz,dmso-d6);hsqc谱(600mhz,dmso-d6)以及hmbc谱(600mhz,dmso-d6)如图8-11所示,未见文献报道,为新化合物,命名为glytacoumestanb。第三化合物:浅黄色固体;hr-esi-msm/z417.16968[m+h]+(calcdforc26h25o5,417.16965);nmr(600mhz,cdcl3)数据见表2;1h-nmr谱(600mhz,cdcl3)、13c-nmr谱(150mhz,cdcl3);hsqc谱(600mhz,cdcl3)以及hmbc谱(600mhz,cdcl3)如图12-15所示,未见文献报道,为新化合物,命名为glytacoumestanc。第四化合物:白色固体;hr-esi-msm/z401.13763[m-h]-(calcdforc25h21o5,401.13835);nmr(600mhz,dmso-d6)数据见表3;1h-nmr谱(600mhz,dmso-d6)、13c-nmr谱(150mhz,dmso-d6);hsqc谱(600mhz,dmso-d6)以及hmbc谱(600mhz,dmso-d6)如图16-19所示,未见文献报道,为新化合物,命名为glytacoumestand。第五化合物:浅色固体;hr-esi-msm/z433.16696[m+h]+(calcdforc26h25o6,433.16456);nmr(600mhz,cdcl3)数据见表3;1h-nmr谱(600mhz,cdcl3)、13c-nmr谱(150mhz,cdcl3);hsqc谱(600mhz,cdcl3)以及hmbc谱(600mhz,cdcl3)如图20-23所示,未见文献报道,为新化合物,命名为glytacoumestane。第五化合物解析:根据hr-esi-ms数据推导出该化合物分子式为c26h26o7,不饱和度为17。1hnmr光谱(表3)显示了两对邻位耦合的芳族质子,分别归属于a环[δh7.86(1h,d,j=9.0hz,h-1),7.00(1h,d,j=9.0hz),h-2)]和b环[δh7.75(1h,d,j=8.4hz,h-7),6.91(1h,d,j=8.4hz,h-8)]。另外,在其13cnmr光谱中观察到了coumestan母核在c-6(δc158.3),c-6a(δc104.1)和c-11a(δc160.3)处的特征碳信号,显示第五化合物为coumestan衍生化合物。通过1hnmr和13cnmr可以观察到两个烯烃双峰信号[δh5.80(1h,d,j=9.6hz),6.87(1h,d,j=9.6hz)],两个甲基信号[δh1.51(6h,s)]和一个氧化的季碳信号(δc77.1),揭示了一个比邻b环的二甲基吡喃环的存在。同时,nmr数据中还揭示了一个2,3-二羟基-3-甲基丁基[δh3.08(1h,dd,j=13.8和10.2hz,ha-1'),3.20(1h,dd,j=13.8和2.4hz,hb-1'),3.71(1h,dd,j=10.2和2.4hz,h-2'),1.37(6h,s,h-4'和h-5');δc26.3(c-1'),78.6(c-2'),73.2(c-3'),26.3(c-4'),24.1(c-5')],一个甲氧基[δh3.98(3h,s),δc56.5]的存在。hmbc谱中,亚甲基质子h-1'与c-3(δc160.2),c-4(δc116.6)和c-4a(δc152.8)具有相关关系,甲氧基质子(δh3.98)与c-3(δc160.2)具有相关关系,揭示两个基团分别在a环的c-4和c-3位置取代。第六化合物:白色固体;hr-esi-msm/z433.16541[m+h]+(calcdforc26h25o6,433.16456);nmr(600mhz,cd3od)数据见表3;1h-nmr谱(600mhz,cd3od)、13c-nmr谱(150mhz,cd3od);hsqc谱(600mhz,cd3od)以及hmbc谱(600mhz,cd3od)如图24-27所示,未见文献报道,为新化合物,命名为glytacoumestanf。第七化合物:淡黄色固体;hr-esi-msm/z419.15118[m+h]+(calcdforc25h23o6,419.14891);nmr(600mhz,cdcl3)数据见表4;1h-nmr谱(600mhz,cdcl3)、13c-nmr谱(150mhz,cdcl3);hsqc谱(600mhz,cdcl3)以及hmbc谱(600mhz,cdcl3)如图28-31所示,未见文献报道,为新化合物,命名为glytacoumestang。第八化合物:淡黄色固体;hr-esi-msm/z367.08096[m-h]-(calcdforc20h15o7,367.08123);nmr(600mhz,cd3od)数据见表4;1h-nmr谱(600mhz,cd3od)、13c-nmr谱(150mhz,cd3od);hsqc谱(600mhz,cd3od)以及hmbc谱(600mhz,cd3od)如图32-35所示,未见文献报道,为新化合物,命名为glytacoumestanh。第九化合物:白色粉末;hr-esi-msm/z353.10162[m-h]-(calcdforc20h17o6,353.10196);nmr(600mhz,cd3od)数据见表4;1h-nmr谱(600mhz,cd3od)、13c-nmr谱(150mhz,cd3od);hsqc谱(600mhz,cd3od)以及hmbc谱(600mhz,cd3od)如图36-39所示,未见文献报道,为新化合物,命名为glytacoumestani。表2本发明提出的新化合物1-3的nmr数据表3本发明提出的新化合物4-6的nmr数据表4本发明提出的新化合物7-9的nmr数据综上所述,本发明提供的一种化合物,发明人通过不断探究发现具有抗类风湿性关节炎功效的新化合物,能够有效抑制lps刺激的一氧化氮的产生,具有较强的体外抗炎活性,同时具有很好的缓解类风湿性关节炎滑膜炎症的能力。本发明提供的一种化合物的制备方法,其通过从烟豆中提取上述化合物,从烟豆复杂的成分组成中提取活性成分。本发明还提供了上述化合物的用途以及以上述化合物作为活性成分的药物,具有很好的市场应用价值。以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1