一种噁噻嗪类化合物及其用途的制作方法
本发明涉及有创新化学药物
技术领域:
,具体是指一种噁噻嗪类化合物及其用途。
背景技术:
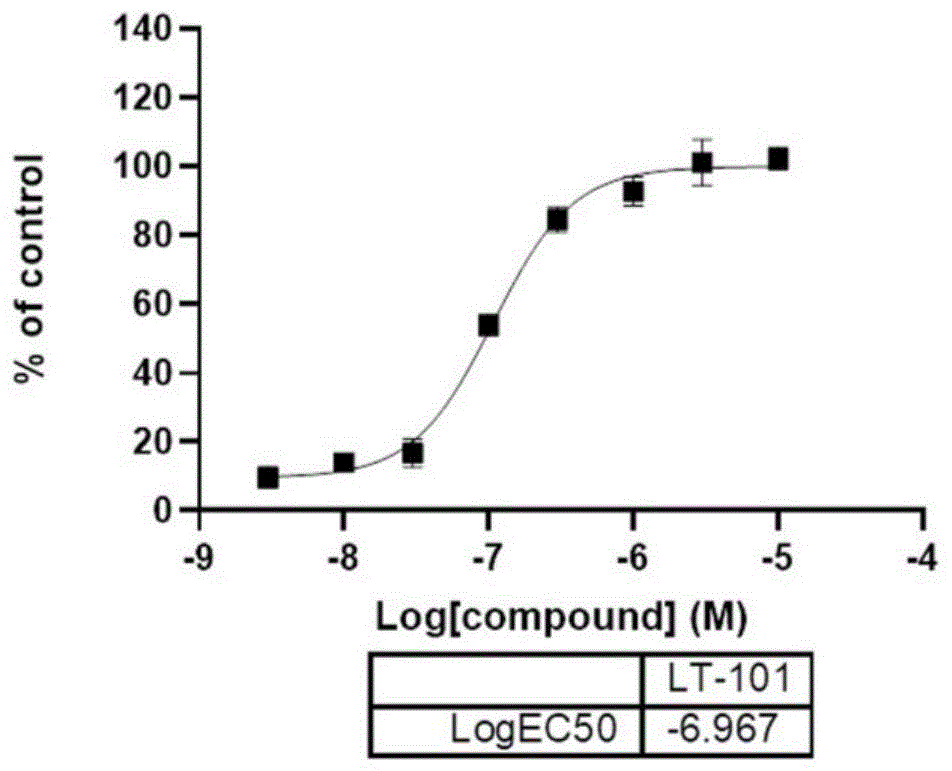
:ampa(α-氨基-3-羟基-5-甲基-4-异噁唑丙酸)受体是离子型谷氨酸受体的重要亚型,主要分布于中枢神经系统的突触后膜,由四种核心亚单位g1ur1~4组成,介导中枢神经系统主要的快速兴奋性传递,促进神经发育和突出可塑性,参与多种神经活动过程,是创新化学药物领域需重点关注的一种与神经系统疾病相关的重要靶点。每个ampa受体亚基的胞外包含两个域:参与亚基组装的氨基末端域(amino-terminaldomain,atd)和提供谷氨酸结合的配体结合域(ligandbindingdomain,lbd)。研究表明,ampa配体结合域(ligandbindingdomain,lbd)位于g1ur2二聚体界面区域,ampa受体的激动剂能够直接与ampa受体的谷氨酸结合位点结合,对其产生作用,但是容易对受体产生过度刺激,引发神经毒性,对大脑产生不可控制的伤害。而ampa受体正向变构调节剂具有与ampa受体激动剂不同的结合位点,可使激动剂(如内源性神经递质谷氨酸)与受体结合后的构象更加稳定,从而降低受体失活率和抑制受体脱敏,使ampa受体的功能得以增强。因此,ampa受体的正向变构调节剂不会出现过度刺激,也不会产生神经毒性的伤害;相比于直接作用于ampa受体的激动剂来说,正向变构调节剂更具优势。随着对ampa受体结构和功能认识的加深,近年来ampa受体正向变构调节剂的发展迅速。ampa受体作为重要的药物靶点,临床基础研究表明ampa受体正向变构调节剂具有脑损伤的神经保护作用、改善认知作用及调控抑郁样行为达到快速抗抑郁作用,被认为是一种治疗神经精神系统疾病有效策略。已有的研究成果为ampa受体正向变构调节剂的设计指明了方向,但是大多处于临床前研究阶段或临床试验阶段。随着构效关系研究的逐渐深入,将更好地帮助人们寻找和发现更多的ampa受体正向变构调节剂,也为以ampa受体为靶标的创新药物研发奠定基础。技术实现要素:本发明的目的在于提供一种结构新颖性,活性较强的噁噻嗪类化合物。本发明另一个目的在于提供上述噁噻嗪类化合物作为ampa受体的正向变构调节剂的具体应用。本发明还有一个目的在于基于上述提供的ampa受体的正向变构调节剂用于制备治疗神经精神系统疾病药物的具体应用。本发明提供一种噁噻嗪类化合物,其通式如下:其中,x为独立的醚键、卤素、r1为独立的h、醚键、或不存在;r2为独立的卤素、甲基、o2s—r5、f2hc—、新丁基、r3为独立的卤素或h;r4为独立的卤素或h;r5为独立的甲基、异丙基;r6为独立的甲基、苯环、苄基、f3c—、异丙基;r7为苯环;r8为h、甲基、醚键、卤素、氰基;r9为独立的醚键、卤素、h;r10为独立的h、卤素、醚键。其包含的具体化合物如下:本发明还提供了一种ampa受体的正向变构调节剂,以上述噁噻嗪类化合物为主要活性成分的生物药学上可接受的盐、多晶型物、溶剂合物。本发明还提供了一种治疗神经精神系统疾病的药物,以上述的ampa受体的正向变构调节剂为主要成分,添加药学上可接受的,对人和动物无毒、无惰性的药用载体和/或赋形剂辅助性成分制备而成的前药或药物组合物。所述的药用载体或赋形剂为一种或多种固体、半固体和液体稀释剂、填料以及药物制品辅剂。所述药物组合物采用制药和食品领域公认的方法制备成各种剂型:喷剂、气雾剂、液体制剂或固体制剂;所述的液体制剂包括注射剂、混悬剂、乳剂、溶液剂或糖浆剂;所述的固体制剂包括片剂、胶囊剂、颗粒剂或冲剂。所述药物治疗的神经精神系统疾病包括精神分裂症、阿尔茨海默病、帕金森病、抑郁症、双相情感障碍。所述药物的给药途径为口服、舌下给药或粘膜透析;所述的注射包括静脉注射、静脉滴注、肌肉注射、腹腔注射或皮下注射。本发明与现有技术相比,具有以下优点及有益效果:本发明合成了一种新型化合物,该化合物能够作为ampa受体的正向变构调节剂,证实了该化合物能够正向调节ampa受体,可使激动剂(如内源性神经递质谷氨酸)与受体结合后的构象更加稳定,从而降低受体失活率和抑制受体脱敏,使ampa受体的功能得以增强,其类药性显著,具有广阔的市场前景。附图说明图1为本发明中化合物lt-101钙内流ec50测试结果曲线图;图2为本发明中化合物lt-103钙内流ec50测试结果曲线图;图3为本发明中化合物lt-129钙内流ec50测试结果曲线图;图4为本发明中化合物lt-134钙内流ec50测试结果曲线图;图5为本发明中化合物lt-134在不加ampa条件下对神经元bdnf表达水平的蛋白质免疫印迹图;图6为本发明中化合物lt-134在不加ampa条件下对神经元bdnf表达水平的统计图;图7为本发明中化合物lt-134在1umampa条件下对神经元bdnf表达水平的蛋白质免疫印迹图;图8为本发明中化合物lt-134在1umampa条件下对神经元bdnf表达水平的统计图;图9为本发明中实验小鼠在化合物lt-134影响下y-迷宫测试结果图;图10为本发明中实验小鼠在化合物lt-134影响下强迫游泳测试结果图。具体实施方式下面结合实施例对本发明作进一步地详细说明,但本发明的实施方式不限于此,在不脱离本发明上述技术思想情况下,根据本领域普通技术知识和惯用手段,做出各种替换和变更,均应包括在本发明的范围内。为使本发明的目的、工艺条件及优点作用更加清楚明白,结合以下实施实例,对本发明作进一步详细说明,此处所描述的具体实施实例仅用以解释本发明,并不用于限定本发明。实施例1:本实施例公开化合物:8-(4-甲氧苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-101)具体合成路线如下:具体制备方法如下:在0℃条件下,将3-溴-2-羟基苯乙酮(a01a)(6.0g,28mmol)分散于60ml二甲基乙酰胺dma中,然后在氮气保护下快速加入氨基磺酰氯(10g,86.5mmol)。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加水析出固体,过滤并分别用水、乙酸乙酯洗涤滤饼,以60%的收率得到化合物为8-溴-4-甲基苯并[e][1,2,3]噁噻嗪2,2-二氧化(a01b)。将a01b(2.75g,10mmol)分散于50mlmeoh溶剂体系里,在室温下分批次加入硼氢化钠(380mg,10.6mmol),加完后继续室温搅拌直到原料a01b反应完全。用饱和氯化铵溶液(20ml)淬灭,并转移至具有30ml乙酸乙酯的分液漏斗中。分离有机层,并用2×30ml乙酸乙酯萃取水层。合并的有机层依次用2×20ml的h2o和1×20ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以85%的收率得到中间体8-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(a01c)。将化合物a01c(55mg,0.2mmol)、对甲氧基苯硼酸a01d(37mg,0.24mmol)、碳酸钾(56mg,0.4mmol)、pd(dppf)cl2(14mg,0.02mmol)、二氧六环(4ml)、h2o(1ml)依次加入25ml两口瓶,氩气保护下80℃搅拌10h。tlc检测反应完全后,加入饱和食盐水20ml,乙酸乙酯(20ml)萃取两次,合并有机相,无水硫酸钠干燥,浓缩,柱色谱分离得到目标产物lt-101,收率83%。ms(esi)306.1[m+h]+;1hnmr(400mhz,dmso)δ8.47(s,1h),7.42–7.33(m,4h),7.29(t,j=7.6hz,1h),7.08–7.02(m,2h),4.81(q,j=6.9hz,1h),3.81(s,3h),1.63(d,j=6.9hz,3h).实施例2:本实施例公开化合物:7-(4-甲氧苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-102)。具体合成路线如下:具体制备方法如下:在0℃条件下,将4-溴-2-羟基苯乙酮(b02a)(6.0g,28mmol)分散于60ml二甲基乙酰胺dma中,然后在氮气保护下快速加入氨基磺酰氯(10g,86.5mmol)。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加水析出固体,过滤并分别用水、乙酸乙酯洗涤滤饼,以63%的收率得到化合物为7-溴-4-甲基苯并[e][1,2,3]噁噻嗪2,2-二氧化(b02b)。将b02b(2.75g,10mmol)分散于50mlmeoh溶剂体系里,在室温下分批次加入硼氢化钠(380mg,10.6mmol),加完后继续室温搅拌直到原料b02b反应完全。用饱和氯化铵溶液(20ml)淬灭,并转移至具有30ml乙酸乙酯的分液漏斗中。分离有机层,并用2×30ml乙酸乙酯萃取水层。合并的有机层依次用2×20ml的h2o和1×20ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以80%的收率得到中间体7-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(b02c)。将化合物b02c(55mg,0.2mmol)、对甲氧基苯硼酸a01d(37mg,0.24mmol)、碳酸钾(56mg,0.4mmol)、pd(dppf)cl2(14mg,0.02mmol)、二氧六环(4ml)、h2o(1ml)依次加入25ml两口瓶,氩气保护下80℃搅拌10h。tlc检测反应完全后,加入饱和食盐水20ml,乙酸乙酯(20ml)萃取两次,合并有机相,无水硫酸钠干燥,浓缩,柱色谱分离得到目标产物lt-102,收率79%。ms(esi)306.1[m+h]+;1hnmr(400mhz,dmso)δ8.46(s,1h),7.66(d,j=8.8hz,2h),7.50(dd,j=8.1,1.7hz,1h),7.44(d,j=8.2hz,1h),7.33(d,j=1.6hz,1h),7.03(d,j=8.8hz,2h),4.79(q,j=6.8hz,1h),3.80(s,3h),1.62(d,j=6.9hz,3h).实施例3:本实施例公开化合物:4-甲基-8-(4-苯氧基苯基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-103)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-苯氧基苯硼酸”。ms(esi)368.1[m+h]+;1hnmr(400mhz,dmso)δ8.51(s,1h),7.49–7.36(m,6h),7.31(t,j=7.6hz,1h),7.19(t,j=7.4hz,1h),7.14–7.04(m,4h),4.83(q,j=6.8hz,1h),1.64(d,j=6.9hz,3h).实施例4:本实施例公开化合物:4-甲基-7-(4-苯氧基苯基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-104)合成路线如实施例2,将“对甲氧基苯硼酸”替换为“4-苯氧基苯硼酸”。ms(esi)368.1[m+h]+;1hnmr(400mhz,dmso)δ8.48(d,j=8.4hz,1h),7.43–7.36(m,3h),7.17–7.01(m,7h),6.87(dd,j=8.6,2.5hz,1h),6.70(d,j=2.5hz,1h),4.74(p,j=7.0hz,1h),1.58(d,j=6.9hz,3h).实施例5:本实施例公开化合物:6-(4-甲氧苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-105)合成路线如下:具体制备方法如下:在0℃条件下,将5-溴-2-羟基苯乙酮(b03a)(6.0g,28mmol)分散于60ml二甲基乙酰胺dma中,然后在氮气保护下快速加入氨基磺酰氯(10g,86.5mmol)。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加水析出固体,过滤并分别用水、乙酸乙酯洗涤滤饼,以60%的收率得到化合物为6-溴-4-甲基苯并[e][1,2,3]噁噻嗪2,2-二氧化(b03b)。将b03b(2.75g,10mmol)分散于50mlmeoh溶剂体系里,在室温下分批次加入硼氢化钠(380mg,10.6mmol),加完后继续室温搅拌直到原料b03b反应完全。用饱和氯化铵溶液(20ml)淬灭,并转移至具有30ml乙酸乙酯的分液漏斗中。分离有机层,并用2×30ml乙酸乙酯萃取水层。合并的有机层依次用2×20ml的h2o和1×20ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以81%的收率得到中间体6-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(b03c)。将化合物b03c(55mg,0.2mmol)、对甲氧基苯硼酸a01d(37mg,0.24mmol)、碳酸钾(56mg,0.4mmol)、pd(dppf)cl2(14mg,0.02mmol)、二氧六环(4ml)、h2o(1ml)依次加入25ml两口瓶,氩气保护下80℃搅拌10h。tlc检测反应完全后,加入饱和食盐水20ml,乙酸乙酯(20ml)萃取两次,合并有机相,无水硫酸钠干燥,浓缩,柱色谱分离得到目标产物lt-105,收率87%。ms(esi)306.1[m+h]+;1hnmr(400mhz,dmso)δ8.47(s,1h),7.70–7.53(m,4h),7.12(d,j=8.3hz,1h),7.03(d,j=8.7hz,2h),4.82(q,j=6.8hz,1h),3.80(s,3h),1.67(d,j=6.9hz,3h).实施例6:本实施例公开化合物:4-甲基-6-(4-苯氧基苯基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-106)合成路线如实施例5,将“对甲氧基苯硼酸”替换为“4-苯氧基苯硼酸”。ms(esi)368.1[m+h]+;1hnmr(400mhz,dmso)δ8.50(s,1h),7.70(d,j=8.7hz,2h),7.67–7.59(m,2h),7.42(t,j=8.0hz,2h),7.21–7.12(m,2h),7.12–7.02(m,4h),4.83(q,j=6.9hz,1h),1.67(d,j=6.9hz,3h).实施例7:本实施例公开化合物:4-甲基-7-(4-苯氧基苯氧基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-107)合成路线如下:具体制备方法如下:将4-苯氧基苯酚(c01a)(334mg,1.8mmol)、4-氟-2-甲氧基苯乙酮(c01b)(336mg,2mmol)和k2co3(552mg,4mmol)溶解到5mldmf溶剂中,将反应体系搅拌慢慢升至120℃,直到反应完全。加入饱和食盐水20ml,乙酸乙酯(20ml)萃取两次,合并有机相,无水硫酸钠干燥,浓缩,柱色谱分离以70%的收率得到2-甲氧基-4-(4-苯氧基苯氧基)苯乙酮(c01c)。在0℃条件下,将c01c(334mg,1mmol)分散于10ml二氯甲烷中,然后在氮气保护下缓慢1ml1m的bbr3四氢呋喃溶液。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加氯化铵水溶液淬灭,并转移至具有20ml乙酸乙酯的分液漏斗中。分离有机层,并用2×20ml乙酸乙酯萃取水层。合并的有机层依次用2×20ml的h2o和1×20ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以86%的收率得到2-羟基-4-(4-苯氧基苯氧基)苯乙酮(c01d)。在0℃条件下,将c01d(160mg,0.5mmol)分散于5ml二甲基乙酰胺dma中,然后在氮气保护下快速加入氨基磺酰氯(175mg,1.5mmol)。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加水析出固体,过滤并分别用水、乙酸乙酯洗涤滤饼,以63%的收率得到4-甲基-7-(4-苯氧基苯氧基)苯并[e][1,2,3]噁噻嗪2,2-二氧化(c01e)。然后将c01e(77mg,0.2mmol)分散于5mlmeoh溶剂体系里,在室温下分批次加入硼氢化钠(10mg,0.2mmol),加完后继续室温搅拌直到原料c01e反应完全。用饱和氯化铵溶液(10ml)淬灭,并转移至具有10ml乙酸乙酯的分液漏斗中。分离有机层,并用2×10ml乙酸乙酯萃取水层。合并的有机层依次用2×10ml的h2o和1×10ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以85%的收率得到4-甲基-7-(4-苯氧基苯氧基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-107)。ms(esi)384.1[m+h]+;1hnmr(400mhz,dmso)δ8.48(d,j=8.4hz,1h),7.42–7.34(m,3h),7.19–7.05(m,5h),7.03(d,j=7.8hz,2h),6.87(dd,j=8.6,2.5hz,1h),6.70(d,j=2.5hz,1h),4.74(p,j=7.0hz,1h),1.58(d,j=6.9hz,3h).实施例8:本实施例公开化合物:7-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-108)合成路线如下:具体制备方法如下:在0℃条件下,将4-溴-2-羟基苯乙酮(b02a)(6.0g,28mmol)分散于60ml二甲基乙酰胺dma中,然后在氮气保护下快速加入氨基磺酰氯(10g,86.5mmol)。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加水析出固体,过滤并分别用水、乙酸乙酯洗涤滤饼,以63%的收率得到化合物为7-溴-4-甲基苯并[e][1,2,3]噁噻嗪2,2-二氧化(b02b)。将b02b(2.75g,10mmol)分散于50mlmeoh溶剂体系里,在室温下分批次加入硼氢化钠(380mg,10.6mmol),加完后继续室温搅拌直到原料b02b反应完全。用饱和氯化铵溶液(20ml)淬灭,并转移至具有30ml乙酸乙酯的分液漏斗中。分离有机层,并用2×30ml乙酸乙酯萃取水层。合并的有机层依次用2×20ml的h2o和1×20ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以80%的收率得到中间体7-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-108)。ms(esi)278.0[m+h]+;1hnmr(400mhz,dmso)δ8.58(d,j=8.3hz,1h),7.44(dd,j=8.3,2.0hz,1h),7.41–7.34(m,2h),4.75(p,j=7.1hz,1h),1.59(d,j=6.9hz,3h).实施例9:本实施例公开化合物:6-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-109)合成路线如实施例8,将“4-溴-2-羟基苯乙酮”替换为“5-溴-2-羟基苯乙酮”。ms(esi)278.0[m+h]+;1hnmr(400mhz,dmso)δ8.56(d,j=8.3hz,1h),7.64–7.62(m,1h),7.57–7.53(m,1h),7.08(d,j=8.7hz,1h),4.78(p,j=7.1hz,1h),1.60(d,j=7.0hz,3h).实施例10:本实施例公开化合物:8-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-110)合成路线如实施例8,将“4-溴-2-羟基苯乙酮”替换为“3-溴-2-羟基苯乙酮”。ms(esi)278.0[m+h]+;1hnmr(400mhz,dmso)δ8.68(s,1h),7.68(d,j=7.8hz,1h),7.43(d,j=7.8hz,1h),7.19(t,j=7.9hz,1h),4.83(q,j=6.9hz,1h),1.61(d,j=6.9hz,3h).实施例11:本实施例公开化合物:8-(4-氯苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-111)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-氯苯硼酸”。ms(esi)310.0[m+h]+;1hnmr(400mhz,dmso)δ8.53(d,j=7.7hz,1h),7.56(d,j=8.5hz,2h),7.45(d,j=8.5hz,3h),7.41–7.37(m,1h),7.33(t,j=7.6hz,1h),4.91–4.77(m,1h),1.64(d,j=6.9hz,3h).实施例12:本实施例公开化合物:8-([1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-112)合成路线如下:具体制备方法如下:在0℃条件下,将3-溴-2-羟基苯乙酮(a01a)(6.0g,28mmol)分散于60ml二甲基乙酰胺dma中,然后在氮气保护下快速加入氨基磺酰氯(10g,86.5mmol)。反应体系搅拌慢慢升至室温,直到反应完全。反应完后加水析出固体,过滤并分别用水、乙酸乙酯洗涤滤饼,以60%的收率得到化合物为8-溴-4-甲基苯并[e][1,2,3]噁噻嗪2,2-二氧化(a01b)。将a01b(2.75g,10mmol)分散于50mlmeoh溶剂体系里,在室温下分批次加入硼氢化钠(380mg,10.6mmol),加完后继续室温搅拌直到原料a01b反应完全。用饱和氯化铵溶液(20ml)淬灭,并转移至具有30ml乙酸乙酯的分液漏斗中。分离有机层,并用2×30ml乙酸乙酯萃取水层。合并的有机层依次用2×20ml的h2o和1×20ml的饱和nacl水溶液洗涤,经na2so4干燥,并在减压下浓缩。通过硅胶色谱法纯化,以85%的收率得到中间体8-溴-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(a01c)。将化合物a01c(550mg,2mmol)、4-氯苯硼酸a11d(375mg,2.4mmol)、碳酸钾(560mg,4mmol)、pd(dppf)cl2(140mg,0.2mmol)、二氧六环(20ml)、h2o(4ml)依次加入100ml两口瓶,氩气保护下80℃搅拌10h。tlc检测反应完全后,加入饱和食盐水20ml,乙酸乙酯(20ml)萃取两次,合并有机相,无水硫酸钠干燥,浓缩,柱色谱分离得到目标产物lt-111,收率80%。将lt-111(61mg,0.2mmol)、苯硼酸a12d(30mg,0.24mmol)、磷酸钾(85mg,0.4mmol)、pd(oac)2(10mg,0.02mmol)、x-phos(20mg,0.04mmol)二氧六环(4ml)、h2o(1ml)依次加入25ml两口瓶,氩气保护下90℃搅拌10h。tlc检测反应完全后,加入饱和食盐水20ml,乙酸乙酯(20ml)萃取两次,合并有机相,无水硫酸钠干燥,浓缩,柱色谱分离得到目标产物lt-112,收率70%。ms(esi)352.1[m+h]+;1hnmr(400mhz,dmso)δ8.54(s,1h),7.79(d,j=8.2hz,2h),7.74(d,j=7.4hz,2h),7.54(d,j=8.1hz,2h),7.50(t,j=7.6hz,2h),7.45(d,j=7.7hz,2h),7.40(t,j=7.3hz,1h),7.36–7.31(m,1h),4.86(q,j=6.5hz,1h),1.66(d,j=6.9hz,3h).实施例13:本实施例公开化合物:8-(6-甲氧基萘-2-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-113)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“6-甲氧基萘-2-硼酸”。ms(esi)356.1[m+h]+;1hnmr(400mhz,dmso)δ8.52(s,1h),7.94–7.85(m,3h),7.53(d,j=8.5hz,1h),7.48(d,j=7.4hz,1h),7.45(d,j=7.5hz,1h),7.38(s,1h),7.37–7.32(m,1h),7.22(dd,j=8.9,2.2hz,1h),4.86(q,j=6.8hz,1h),3.91(s,3h),1.66(d,j=6.9hz,3h).实施例14:本实施例公开化合物:8-(2,3-二氢苯并[b][1,4]二噁英-6-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-114)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“苯并-1,4-二氧六环-6-硼酸”。ms(esi)334.1[m+h]+;1hnmr(400mhz,dmso)δ8.48(s,1h),7.38(d,j=7.6hz,1h),7.35–7.33(m,1h),7.27(t,j=7.6hz,1h),6.96(d,j=8.3hz,1h),6.93–6.88(m,2h),4.81(q,j=6.7hz,1h),4.29(s,4h),1.63(d,j=6.9hz,3h).实施例15:本实施例公开化合物:8-(3-氟-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-115)合成路线如实施例12,将“4-氯苯硼酸”替换为“4-氯-2-氟苯硼酸”。ms(esi)370.1[m+h]+;1hnmr(400mhz,dmso)δ8.59(s,1h),7.67(d,j=8.5hz,1h),7.63(d,j=7.1hz,2h),7.54–7.33(m,8h),4.86(q,j=6.8hz,1h),1.66(d,j=6.9hz,3h).实施例16:本实施例公开化合物:4-甲基-8-(4-(三氟甲氧基)苯基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-116)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-三氟甲氧基苯硼酸”。ms(esi)360.0[m+h]+;1hnmr(400mhz,dmso)δ8.56(s,1h),7.56(d,j=8.7hz,2h),7.50(d,j=8.4hz,2h),7.46(d,j=7.8hz,1h),7.42(d,j=6.5hz,1h),7.34(t,j=7.6hz,1h),4.85(q,j=6.8hz,1h),1.65(d,j=6.9hz,3h).实施例17:本实施例公开化合物:4-甲基-8-(4-甲苯基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-117)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-甲苯硼酸”。ms(esi)290.1[m+h]+;1hnmr(400mhz,dmso)δ8.47(s,1h),7.40(d,j=7.5hz,1h),7.35(d,j=6.1hz,1h),7.30(q,j=8.1hz,5h),4.82(q,j=6.8hz,1h),2.36(s,3h),1.63(d,j=6.9hz,3h).实施例18:本实施例公开化合物:8-(4-异丙氧基苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-118)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-异丙氧基苯硼酸”。ms(esi)334.1[m+h]+;1hnmr(400mhz,dmso)δ8.47(s,1h),7.40–7.32(m,4h),7.28(t,j=7.6hz,1h),7.01(d,j=8.7hz,2h),4.81(q,j=6.8hz,1h),4.67(hept,j=6.0hz,1h),1.63(d,j=6.9hz,3h),1.30(d,j=6.0hz,6h).实施例19:本实施例公开化合物:4-甲基-8-(4-(甲磺酰)苯基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-119)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-(甲磺酰基)苯硼酸”。ms(esi)354.0[m+h]+;1hnmr(400mhz,dmso)δ8.60(s,1h),8.04(d,j=8.3hz,2h),7.70(d,j=8.3hz,2h),7.51(d,j=7.5hz,1h),7.45(d,j=6.8hz,1h),7.37(t,j=7.7hz,1h),4.86(q,j=6.8hz,1h),3.29(s,3h),1.65(d,j=6.9hz,3h).实施例20:8-(3-氟-4-甲氧苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-120)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“3-氟-4-甲氧苯硼酸”。ms(esi)324.0[m+h]+;1hnmr(400mhz,dmso)δ8.52(s,1h),7.40(t,j=7.2hz,2h),7.33–7.20(m,4h),4.82(q,j=6.8hz,1h),3.90(s,3h),1.63(d,j=6.9hz,3h).实施例21:本实施例公开化合物:8-(4-氟苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-121)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-氟苯硼酸”。ms(esi)294.0[m+h]+;1hnmr(400mhz,dmso)δ8.52(s,1h),7.51–7.45(m,2h),7.45–7.42(m,1h),7.40–7.37(m,1h),7.36–7.29(m,3h),4.83(q,j=6.9hz,1h),1.64(d,j=6.9hz,3h).实施例22:本实施例公开化合物:8-(4-(异丙基磺酰)苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-122)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-(异丙基磺酰)苯硼酸”。ms(esi)382.1[m+h]+;1hnmr(400mhz,dmso)δ8.59(s,1h),7.97(d,j=8.3hz,2h),7.72(d,j=8.4hz,2h),7.51(d,j=7.6hz,1h),7.47(d,j=6.6hz,1h),7.37(t,j=7.7hz,1h),4.86(q,j=6.8hz,1h),3.57–3.44(m,1h),1.65(d,j=6.9hz,3h),1.21(d,j=6.8hz,6h).实施例23:本实施例公开化合物:8-(4-(苄氧基)苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-123)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-苄氧基苯硼酸”。ms(esi)382.1[m+h]+;1hnmr(400mhz,dmso)δ8.47(s,1h),7.48(d,j=7.4hz,2h),7.44–7.32(m,7h),7.29(t,j=7.6hz,1h),7.12(d,j=8.7hz,2h),5.16(s,2h),4.81(q,j=6.8hz,1h),1.63(d,j=6.9hz,3h).实施例24:本实施例公开化合物:(4-(4-甲基-2,2-二羟基-3,4-二氢苯并[e][1,2,3]氧杂噻嗪-8-基)苯基)(苯基)甲酮(lt-124)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-苯甲酰苯硼酸”。ms(esi)380.1[m+h]+;1hnmr(400mhz,dmso)δ8.58(s,1h),7.86(d,j=8.3hz,2h),7.82–7.78(m,2h),7.70(t,j=7.4hz,1h),7.63(d,j=8.3hz,2h),7.59(t,j=7.6hz,2h),7.50(d,j=7.7hz,1h),7.47(d,j=7.6hz,1h),7.37(t,j=7.7hz,1h),4.86(q,j=6.8hz,1h),1.66(d,j=6.9hz,3h).实施例25:本实施例公开化合物:8-(苯并[d][1,3]二氧杂环戊烯-5-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-125)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“3,4-亚甲二氧基苯硼酸”。ms(esi)320.0[m+h]+;1hnmr(400mhz,dmso)δ8.48(s,1h),7.39(d,j=7.6hz,1h),7.37–7.33(m,1h),7.28(t,j=7.6hz,1h),7.03(d,j=8.0hz,1h),6.95(d,j=1.6hz,1h),6.90(dd,j=8.0,1.7hz,1h),6.08(s,2h),4.81(q,j=6.9hz,1h),1.63(d,j=6.9hz,3h).实施例26:本实施例公开化合物:8-(4-(二氟甲基)苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-126)合成路线如实施例23,“2-羟基-3-甲氧基苯甲醛”替换为“3-溴-2-羟基苯甲醛”ms(esi)263.9[m+h]+;1hnmr(400mhz,dmso)δ8.75(s,1h),7.67(d,j=7.8hz,1h),7.35(t,j=11.4hz,1h),7.17(t,j=7.8hz,1h),4.62(s,2h).实施例27:本实施例公开化合物:8-(4-(叔-丁基)苯基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-127)合成路线如实施例1,将“对甲氧基苯硼酸”替换为“4-叔丁基苯硼酸”。ms(esi)332.1[m+h]+;1hnmr(400mhz,dmso)δ8.50(s,1h),7.51(d,j=8.4hz,2h),7.43–7.35(m,4h),7.30(t,j=7.6hz,1h),4.82(q,j=6.8hz,1h),1.64(d,j=6.9hz,3h),1.33(s,9h).实施例28:本实施例公开化合物:4-甲基-8-(4'-甲基-[1,1'-联苯基]-4-基)-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-128)合成路线如实施例12,将“苯硼酸”替换为“4-甲基苯硼酸”。ms(esi)366.1[m+h]+;1hnmr(400mhz,dmso)δ8.54(s,1h),7.76(d,j=8.3hz,2h),7.64(d,j=8.1hz,2h),7.52(d,j=8.2hz,2h),7.44(d,j=8.2hz,2h),7.37–7.32(m,1h),7.30(d,j=8.0hz,2h),4.85(q,j=6.8hz,1h),2.36(s,3h),1.65(d,j=6.9hz,3h).实施例29:本实施例公开化合物:8-(4'-甲氧基-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-129)合成路线如实施例12,将“苯硼酸”替换为“4-甲氧基苯硼酸”。ms(esi)382.1[m+h]+;1hnmr(400mhz,dmso)δ8.53(s,1h),7.74(d,j=8.3hz,2h),7.69(d,j=8.7hz,2h),7.50(d,j=8.3hz,2h),7.43(d,j=7.8hz,2h),7.37–7.28(m,1h),7.05(d,j=8.7hz,2h),4.84(q,j=6.9hz,1h),3.81(s,3h),1.65(d,j=6.9hz,3h).实施例30:本实施例公开化合物:8-(3'-甲氧基-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-130)合成路线如实施例12,将“苯硼酸”替换为“3-甲氧基苯硼酸”。ms(esi)382.1[m+h]+;1hnmr(400mhz,dmso)δ8.54(d,j=8.4hz,1h),7.80(d,j=8.3hz,2h),7.53(d,j=8.3hz,2h),7.45(d,j=8.2hz,2h),7.43–7.38(m,1h),7.35(t,j=7.7hz,1h),7.32–7.25(m,2h),6.97(dd,j=8.1,2.0hz,1h),4.85(p,j=7.0hz,1h),3.84(s,3h),1.66(d,j=6.9hz,3h).实施例31:本实施例公开化合物:8-(2'-甲氧基-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-131)合成路线如实施例12,将“苯硼酸”替换为“2-甲氧基苯硼酸”。ms(esi)382.1[m+h]+;1hnmr(400mhz,dmso)δ8.55(s,1h),7.59(d,j=8.2hz,2h),7.47(d,j=8.2hz,2h),7.44(d,j=7.7hz,2h),7.40–7.30(m,3h),7.14(d,j=8.2hz,1h),7.06(t,j=7.3hz,1h),4.85(q,j=6.7hz,1h),3.80(s,3h),1.66(d,j=6.9hz,3h).实施例32:本实施例公开化合物:8-(3',4'-二氟-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-132)合成路线如实施例12,将“苯硼酸”替换为“3,4-二氟苯硼酸”。ms(esi)370.1[m+h]+;1hnmr(400mhz,dmso)δ8.55(d,j=8.3hz,1h),7.90–7.84(m,1h),7.82(d,j=8.3hz,2h),7.68–7.60(m,1h),7.59–7.50(m,3h),7.48–7.41(m,2h),7.35(t,j=7.6hz,1h),4.85(p,j=6.9hz,1h),1.66(d,j=6.9hz,3h).实施例33:本实施例公开化合物:8-(4'-氟-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-133)合成路线如实施例12,将“苯硼酸”替换为“4-氟苯硼酸”。ms(esi)370.1[m+h]+;1hnmr(400mhz,dmso)δ8.55(s,1h),7.83–7.74(m,4h),7.53(d,j=8.3hz,2h),7.47–7.42(m,2h),7.38–7.27(m,3h),4.85(q,j=6.7hz,1h),1.66(d,j=6.9hz,3h).实施例34:本实施例公开化合物:8-(2'-氟-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-134)合成路线如实施例12,将“苯硼酸”替换为“2-氟苯硼酸”。ms(esi)370.1[m+h]+;1hnmr(400mhz,dmso)δ8.56(d,j=8.4hz,1h),7.68(d,j=7.0hz,2h),7.62(td,j=7.8,1.4hz,1h),7.55(d,j=8.2hz,2h),7.49–7.42(m,3h),7.39–7.30(m,3h),4.86(p,j=7.0hz,1h),1.66(d,j=6.9hz,3h).实施例35:本实施例公开化合物:8-(3'-氟-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-135)合成路线如实施例12,将“苯硼酸”替换为“3-氟苯硼酸”。ms(esi)370.1[m+h]+;1hnmr(400mhz,dmso)δ8.55(d,j=8.3hz,1h),7.84(d,j=8.3hz,2h),7.64–7.58(m,2h),7.57–7.49(m,3h),7.45(dd,j=7.7,2.2hz,2h),7.35(t,j=7.6hz,1h),7.26–7.19(m,1h),4.85(p,j=6.9hz,1h),1.66(d,j=6.9hz,3h).实施例36:本实施例公开化合物:8-(4'-氰基-[1,1'-联苯基]-4-基)-4-甲基-3,4-二氢苯并[e][1,2,3]噁噻嗪2,2-二氧化(lt-136)合成路线如实施例12,将“苯硼酸”替换为“4-氰基苯硼酸”。ms(esi)399.1[m+h]+;1hnmr(400mhz,dmso)δ8.56(s,1h),8.01–7.93(m,4h),7.89(d,j=8.3hz,2h),7.59(d,j=8.3hz,2h),7.49–7.43(m,2h),7.35(t,j=7.6hz,1h),4.85(q,j=6.7hz,1h),1.66(d,j=6.9hz,3h).实施例37:本实施例以上述实施例提供的36个化合物为基础,对其进行生物活性测试实验,具体如下:制备大鼠原代神经元:神经元取材于孕19天的sd大鼠,将胚胎置于预冷的hbss缓冲液中。显微镜下取出胎鼠大脑,去除脑膜,分离海马,用虹膜剪剪碎脑组织成1mm3大小糊状,加入含10%fbs的dmem培液,吹散制备成单细胞悬液,以2×105/ml接种于pdl包被好的培养板中,置于培养箱中培养。6h后换液,以neurobasal+2%b27+2mm谷氨酰胺培养基继续培养,每2天换液。进行ca2+内流检测:原代神经元培养5天后进行ca2+内流的小分子筛选。吸去培养基后,每孔加入100μlloadingbuffer(3μm荧光钙指示剂fluo4-am、0.04%pluronicf-127、1.25mm丙磺舒、10mmhepes、0.05%bsa、dmem),并将细胞在培养箱孵育50分钟。然后吸去loadingbuffer,用recordingmedium(1.25mm丙磺舒、10mmhepes、0.05%bsa、dmem)洗涤一次,再次向每个孔中添加100μlrecordingmedium。使用荧光酶标仪检测在有和无5μms-ampa的情况下化合物诱导的细胞内ca2+水平的相对增加,持续15分钟。化合物的活性定义为在整个测量期间积分的荧光强度。强度0%定义为仅存在5μms-ampa时的活性,而强度100%定义为存在5μms-ampa和10μmtak-137时的活性。活性(%)=(m-a)/(n-a),m:1μm的待测化合物在加入5μms-ampa后引起荧光的增量;n:10μm阳性化合物tak-137在加入5μms-ampa后引起荧光的增量;a:只加入5μms-ampa后引起荧光的增量。钙内流测试结果如表一所示(结果为3次及以上重复试验平均值):表一全部化合物钙内流测试结果根据上表,选取化合物lt-101、lt-103、lt-129、lt-134进行钙内流ec50测试。使用逻辑回归分析计算ec50值,数据均为3次及以上重复试验平均值。数据处理采用graphpad分析。体内药效数据表示为mean±sem(n=6-10每组),两组间比较采用独立样本t检验(unpairedstudent'st-test),多组间比较采用单因素方差分析(one-wayanova)统计,p<0.05认为有统计学差异(*p<0.05,**p<0.01,***p<0.001,****p<0.0001)。具体结果,如图1~4所示,具体数值见如表二所示表二部分化合物钙内流ec50测试化合物编号logec50(m)化合物编号logec50(m)lt-101-6.967lt-129-7.032lt-103-6.635lt-134-7.416实验结论:噁噻嗪类衍生物能够剂量依赖的促进原代神经元钙离子内流,对ampa受体表现出正向调节活性。实施例89:本实施例重点针对化合物lt-134进行神经元脑源性神经营养因子(bdnf)表达水平的检测。具体检测过程如下:从孕19天的sd大鼠胚胎中分离皮层,用眼科剪将皮层剪成1mm*1mm的组织块,加入含10%fbs的dmem培养基,均匀机械吹打成散细胞,经细胞筛过滤成为单细胞悬液,以3*105/cm2密度种于6孔板,培养24h后,换成含2%b27的neurobasal培养基,每隔两天换液,培养至第5天,在加入或不加入1μmampa的情况下,将各浓度(62.5nm、125nm、250nm、500nm)的lt-134化合物加入6孔板培养24h,随后收集蛋白样品,进行后续bdnf的蛋白表达水平分析。测试结果如图5~8所示,根据图5~8可知,在不加ampa的条件下,化合物lt-134不显著影响神经元bdnf的表达(图5,图6),在加入1μmampa后,化合物lt-134能够剂量依赖性的增加神经元bdnf的蛋白表达水平(图7,图8)。实施例90:本实施例针对化合物lt-134对其进行体内药效实验,具体实验如下:4.1实验动物c57小鼠,雄性,20-25g;购自成都达硕实验动物有限公司,本单位动物实验室饲养。饲养条件:温度25±1℃,湿度50-60%,12/12h昼夜节律,标准动物饲养喂养,实验前在动物实验室适应环境7天,自由进食、饮水。4.2药品测试化合物加入终体积1%dmso、4%吐温80和95%生理盐水,涡旋或超声使充分混匀。mk-801用生理盐水溶于生理盐水(0.9%氯化钠)溶液。小鼠腹腔注射,所有的药物小鼠给药体积分别为10ml/kg。4.3实验方案动物模型:腹腔注射mk-801(0.2mg/kg)构建精神分裂症的小鼠模型给药方案:正常小鼠腹腔给药生理盐水;模型小鼠腹腔给药mk-801(0.2mg/kg);治疗小鼠先腹腔给药lt-134(0.1mg/kg)半小时后再腹腔mk-801(0.2mg/kg);治疗小鼠腹腔给药lt-134(0.1和0.3mg/kg)。y迷宫测试:由3等长臂组成(31cm×9cm×16cm),每两个臂之间夹角为120度,在中央处各有一个可移动的隔板,将c57小鼠放在y迷宫任意一臂末端,任其自由探索5min,摄像系统记录动物的行为变化。记录以下各项指标:动物进入迷宫臂的次数(以小鼠四只脚均进入臂为进臂一次标准);轮流一次次数(依次连续进入y迷宫全部三个臂一次)。通过计算小鼠正确轮流百分比来评价其工作记忆。alternationindex=正确轮流次数/总轮流次数。测试结果如图9所示,根据图9显示结果可知,化合物lt-134能够显著提高mk-801造模小鼠的工作记忆功能。强迫游泳测试:实验装置是一个直径15厘米,高25厘米的透明的有机塑料圆桶,水深15厘米,水温控制在25±1℃。总测试6分钟,前两分钟为适应过程,记录后四分钟小鼠不动时间。小鼠不动的定义是:小鼠头朝上漂浮在水面或为保持头部浮在水面而做必要的小幅游动。测试结果如图10所示,根据图10显示结果可知,化合物lt-134在0.1和0.3mg/kg的剂量下都能够达到快速抗抑郁。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1