一种聚硅醚及钴催化潜手性硅烷与二醇的选择性脱氢偶联合成聚硅醚的方法
1.本发明属于含硅高分子合成技术领域,具体涉及一种聚硅醚及应用钴的双膦配体配合物催化合成结构新颖的聚硅醚的方法。
背景技术:
2.由于具有多样的性质以及地壳中硅和氧的储量丰富,主链含有硅氧键的聚合物一直是研究热点(参考文献一:w.m.haynes,abundance of elements in the earth’s crust andin the sea,crc handbook of chemistry and physics,96th ed.;crcpress,2015-2016;sections14-18.)。到目前为止,一些主链含有硅氧键的聚合物逐渐被发展和应用,其中包括聚硅氧烷、聚硅醚和聚硅酯等。由于主链结构的相似性,这些聚合物常常表现出类似的性质,如较低的玻璃化转变温度,良好的热稳定性、生物兼容性、透气性以及可降解性等(参考文献二:y.kawakami,y.li,des.monomers polym.2000,3,399.)。作为其中的一个成功的例子,聚二甲基硅氧烷具有良好的柔韧性以及热稳定性,经过几十年的发展,其已被广泛应用于硅油、弹性体、粘合剂以及表面材料等领域。然而聚二甲基硅氧烷的机械性能往往较差,这也影响其进一步应用(参考文献三:j.m.zeigler,f.w.g.fearon,in silicon based polymer science:acomprehensive resource;acs press:washington,dc,1990;vol.224,pp 265-281.)。
3.从重复单元结构上来看,聚硅醚具有聚硅氧烷和聚碳硅烷的特点。这也决定了聚硅醚与聚硅氧烷相比机械性质会更好,可调性也更多。正是因此,关于聚硅醚的研究逐渐兴起。在研究初期,聚硅醚的常常是通过二醇与氯硅烷或氮硅烷的聚缩合反应来合成(参考文献四:a)w.r.dunnavant,r.a.markle,r.g.sinclair,p.b.stickney,j.e.curry,j.d.byrd,macromolecules1968,1,249.b)k.drake,i.mukherjee,k.mirza,h.-f.ji,j.-c.bradley,y.wei,macromolecules2013,46,4370.(c)y.imai,j.macromol.sci.,chem.1991,28,1115.)。为得到高分子量的聚合产物,反应中需要加入当量的碱或使用减压条件。但该方法的弊端在于反应的原子经济性较差,耗能较高。这也限制着该聚合反应的发展。之后,nishikubo小组利用tpbc催化环氧与氯硅烷的加成聚合反应合成了一系列主链上带有氯甲基的聚硅醚(参考文献五:a)t.nishikubo,a.kameyama,y.kimura,k.fukuyo,macromolecules1995,28,4361.b)t.nishikubo,a.kameyama,y.kimura,t.nakamura,macromolecules1996,29,5529.)。该反应条件温和,原子经济性高,但所使用的的氯硅烷较为不稳定。为了替换不稳定的氯硅烷,人们逐渐将硅烷与醇的脱氢偶联以及硅烷与醛或酮的硅氢化反应应用于聚硅醚的合成(参考文献六:a)y.kawakami,i.imae,acs symp.ser.2003,838,61.b)j.m.mabry,m.k.runyon,w.p.weber,macromolecules2002,35,2207.c)g.l
á
zaro,m.iglesias,f.j.fern
á
ndez-alvarez,p.j.sanz miguel,j.j.p
é
rez-torrente,l.a.oro,chemcatchem2013,5,1133.)。
4.作为一种高效且原子经济性的反应,脱氢偶联聚合已被应用于聚硅醚的合成。而
基于钌、铑、钯、铂等贵金属的脱氢偶联催化体系也逐渐发展完善(参考文献七:a)y.li,y.kawakami,macromolecules1999,32,8768.b)y.li,y.kawakami,macromolecules1999,32,3540.c)y.li,y.kawakami,macromolecules1999,32,6871.d)r.zhang,j.e.mark,a.r.pinhas,macromolecules2000,33,3508.e)m.oishi,j.-y.moon.w.janvikul,y.kawakami,poly int.2001,50,135.f)y.li,m.seino,y.kawakami,macromolecules2000,33,5311.)。尽管取得了很大进展,然而上述基于贵金属的催化体系的缺点也变得越来越明显,例如贵金属催化剂的高成本、低储量、高催化剂用量以及生物兼容性等问题。因此,发展基于廉价金属铁、钴、锰的催化体系更有利于解决以上问题。最近,基于铁和钴的脱氢偶联催化体系已被报道(参考文献八:a)c.lichtenberg,l.viciu,m.adelhardt,j.sutter,k.meyer,b.de bruin,h.gru tzmacher,angew.chem.int.ed.2015,54,5766.b)s.vijjamarri,s.streed,e.m.serum,m.p.sibi,g.du,acs sustainable chem.eng.2018,6,2491.)。然而,钴催化的醇与硅烷的脱氢偶联聚合反应还未被发展起来。此外,影响聚硅醚发展另一个问题是目前所合成的聚硅醚结构较为类似,这不利于对聚硅醚的性质进行调节。在2000年,kawakami小组报道了一种铑催化的潜手性硅烷与二醇的选择性脱氢偶联,得到了一系列主链上含有硅氢基团的结构新颖的聚硅醚(参考文献九:y.li,m.seino,y.kawakami,macromolecules2000,33,5311.)。这种聚硅醚所含有的硅氢基团可能与水或烯烃反应,进而可被用于手性柱填料领域。基于以上文献背景的调研,在此我们报道一种钴催化的选择性脱氢偶联反应,该方法以潜手性硅烷与二醇为原料合成主链上含有硅氢基团的结构新颖的聚硅醚。
技术实现要素:
5.本发明的目的是提供一种聚硅醚及合成聚硅醚的方法,是以钴与双膦配体的配合物为催化剂,催化含有羟基硅烷单体(ab型或aa和bb型)的选择性脱氢偶联聚合反应制备主链上含有硅氢基团的聚硅醚的方法,实现不同类型单体脱氢偶联聚合。
6.为实现上述目的,本发明采用的技术方案如下:
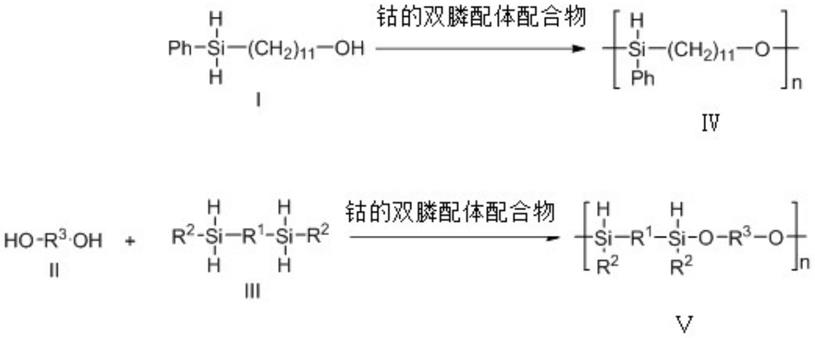
7.一种聚硅醚,具有如通式iv或v所示的结构:
[0008][0009]
式中:r1和r2均为烷基或芳基;r3为c
6-12
的烷基。
[0010]
基于以上技术方案,优选地,所述r1和r2的烷基优选为c
2-8
的烷基。
[0011]
一种钴催化潜手性硅烷与二醇的选择性脱氢偶联合成聚硅醚的方法,以钴的双膦配体配合物为催化剂,催化单体ⅰ选择性脱氢偶联聚合制备所述聚硅醚,或催化单体ⅱ和ⅲ脱氢偶联聚合制备所述聚硅醚;
[0012]
所述钴的双膦配体配合物与底物的摩尔比为:0.005~0.020:1;当使用为单体ⅰ为底物时,所述钴的双膦配体配合物与单体ⅰ的摩尔比均为:0.005~0.020:1;当使用单体ⅱ和单体ⅲ为底物时,所述钴的双膦配体配合物与单体ⅱ的摩尔比为:0.005~0.020:1,优选为0.010~0.020:1,且所述单体ⅱ与单体ⅲ的摩尔比为1:1~1:1.2,优选为1:1;
[0013]
所述含有羟基硅烷的ab型单体结构式为式ⅰ,aa和bb型单体的结构式分别为式ⅱ和式ⅲ,具体的反应式如下:
[0014][0015]
式中:r1和r2均为烷基或芳基;r3为c
6-12
的烷基;
[0016]
反应温度:40~120℃;反应时间12-48小时。
[0017]
基于以上技术方案,优选地,所述r1和r2的烷基优选为c
2-8
的烷基。基于以上技术方案,优选地,当使用手性双膦配体时,反应能够得到手性聚硅醚。
[0018]
基于以上技术方案,优选的,所述反应温度为60~100℃。
[0019]
基于以上技术方案,优选的,所述合成聚硅醚的方法步骤如下:
[0020]
在氮气保护下,将单体ⅰ加入所述催化剂溶液中,于40~120℃反应12-48小时,得到所述聚硅醚;或在氮气保护下,将单体ⅱ和单体ⅲ加入所述催化剂溶液中,于40~120℃反应12-48小时,得到所述聚硅醚;
[0021]
其中,所述催化剂溶液为所述催化剂溶于有机溶剂形成。
[0022]
基于以上技术方案,优选地,所述脱氢偶联反应中使用的有机溶剂选自均三甲苯(mesitylene)、叔丁基甲醚(
t
buome)、乙腈(mecn)、四氢呋喃(thf)、正己烷(
n
hexane)中的至少一种,优选正己烷。
[0023]
基于以上技术方案,优选地,所述钴的双膦配体配合物是通过钴的前驱体与双膦配体在20-40℃下有机溶剂中搅拌反应再去除有机溶剂得到。
[0024]
基于以上技术方案,优选地,所述钴的前驱体为乙酰丙酮钴(ⅱ)co(acac)2、乙酰丙酮钴(ⅲ)co(acac)3、乙酸钴co(oac)2中的至少一种,优选为乙酰丙酮钴(ⅱ)co(acac)2。
[0025]
基于以上技术方案,优选地,所述钴的前驱体与双膦配体的摩尔比为1:1.0至1:1.2,优选为1:1.0。
[0026]
基于以上技术方案,优选地,所述钴的双膦配体配合物所使用的有机溶剂选自叔丁基甲醚(
t
buome)、甲苯(toluene)、四氢呋喃(thf)中的至少一种,优选为四氢呋喃。
[0027]
基于以上技术方案,优选地,所述双膦配体为1,4-双(二苯基膦)丁烷,1,5-双(二苯基膦)戊烷,1,6-双(二苯基膦)己烷,1,1'-联萘-2,2'-双二苯膦,4,5-双二苯基膦-9,9-二甲基氧杂蒽或(s)-(-)-(6,6'-二甲氧基联苯-2,2'-基)双(二苯基膦)。
[0028]
基于以上技术方案,优选地,所述单体ⅰ在所述催化剂溶液的的浓度为0.1~1.0mmol/ml,或所述单体ⅱ和单体ⅲ在所述催化剂溶液的浓度分别为0.1~1.0mmol/ml。
[0029]
作为优选的技术方案,所述合成聚硅醚方法通过两步一锅法制备,即为将合成单体ⅲ的底物加入催化剂溶液中,通过硅氢化反应得到单体ⅲ,再加入单体ⅱ,继续反应得到
聚硅醚。具体步骤如下:
[0030]
(1)催化剂制备:将钴的前驱体与双膦配体在有机溶剂中搅拌反应,得到催化剂溶液;
[0031]
(2)合成聚硅醚:在氮气保护下,将合成单体ⅲ的底物加入催化剂溶液中,通过硅氢化反应得到单体ⅲ,随后加入单体ⅱ,继续反应得到聚硅醚。例如合成单体ⅲ的底物可以为1,5-己二烯和苯基硅烷。
[0032]
基于以上技术方案,优选地,步骤(2)中,待反应结束,除去有机溶剂后加入二氯甲烷溶解产物,滴加冷甲醇使产物析出,去除上层有机溶剂,抽干得聚合产物聚硅醚。
[0033]
基于以上技术方案,优选地,步骤(2)中,除去溶剂的方法为减压除去溶剂。
[0034]
本发明的有益效果:
[0035]
本发明以廉价金属钴的双膦配体配合物作为催化剂,通过催化潜手性硅烷与二醇的选择性脱氢偶联得到一系列结构新颖的聚硅醚。所合成的聚硅醚来源于不同类型的单体。此外,该催化剂还能实现硅氢化/脱氢偶联两步一锅法合成聚硅醚。当使用手性双膦配体时,反应能够得到手性聚硅醚。本发明具有以下优点:
[0036]
1.聚硅醚的数均分子量最高可达到3.23
×
104;
[0037]
2.催化剂制备方便,反应操作简便实用,反应条件温和;
[0038]
3.聚硅醚的主链上含有硅氢基团,结构新颖;
[0039]
4.合成的聚硅醚耐高温性能良好;
[0040]
5.采用的两步一锅法操作步骤简单,高效;
[0041]
6.合成的手性聚硅醚在手性分离领域具有潜在的实际应用价值。
具体实施方式
[0042]
下面通过实施例详述本发明,但本发明并不限于下述的实施例。
[0043]
本文单独或组合使用的术语“烷基”是指任选取代的直链或任选取代的支链的饱和脂肪族烃类。本发明的“烷基”优选可具有1-约20个碳原子,例如具有1-约10个碳原子,或1-约8个碳原子,不限于甲基、乙基、正丙基、异丙基、2-甲基-1-丙基、2-甲基-2-丙基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基小戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、4-甲基-2-戊基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、叔戊基和己基,如庚基和辛基等。本文定义的基团如“烷基”出现数字范围时,例如“c
2-c6烷基”或“c
2-6
烷基”是指可由2个碳原子、3个碳原子、4个碳原子、5个碳原子或6个碳原子构成的烷基,本文的烷基也包括未指定数字范围的情况。
[0044]
本文单独或组合使用的术语“芳香基/芳基”是指任取代的芳香烃基,其具有6-约20个,如6-12个或6-10个成环碳原子,其可以是单环芳基、双环芳基或更多环芳基。双环芳基或更多环芳基可以是一个单环芳基与其它独立环,如脂环、杂环、芳环、芳杂环或更多环芳基。单环芳基的非限定性实施例包括6至约12个、6至约10个或6至约8个成环碳原子的单环芳基,例如苯基;双环芳基例如奈基;多环芳基例如菲基、蒽基、薁基。
[0045]
钴的双膦配体配合物是通过乙酰丙酮钴co(acac)2与双膦配体在四氢呋喃中搅拌反应五分钟再抽干溶剂得到,所用原料为乙酰丙酮钴co(acac)2(cas:14024-48-7),1,4-双
(二苯基膦)丁烷dppb(cas:7688-25-7),1,5-双(二苯基膦)戊烷dpppe(cas:27721-02-4),1,6-双(二苯基膦)己烷dppph(cas:19845-69-3),1,1'-联萘-2,2'-双二苯膦(cas:98327-87-8)4,5-双二苯基膦-9,9-二甲基氧杂蒽(cas:161265-03-8),(s)-(-)-(6,6'-二甲氧基联苯-2,2'-基)双(二苯基膦)(cas:133545-17-2)均为市售且无需任何处理。
[0046]
下述实施例中二醇7a-f是通过用乙酸乙酯重结晶得到。
[0047]
单体3b是参考文献(参考文献十:y.li,m.seino,y.kawakami,macromolecules2000,33,15.)合成;3c是参考文献(参考文献十一:y.li,m.seino,y.kawakami,macromolecules 2000,33,15.(a)l.xue,y.kawakami,polym.j.1993,25,1003.(b)t.imori,h.woo,j.f.walzer,t.d.tilley,chem.mater.1993,5,1487.)合成。第一步是格式反应,第二步是氢化铝锂还原得到单体。
[0048]
实施例1
[0049]
单体3a的合成
[0050][0051]
根据改进的文献方法(参考文献十二:c.wang,w.-j.teo,s.ge,acs catal.2017,7,855.)可以方便地制备1,6-双(苯基硅烷基)己烷(3a)。氮气保护下,在反应瓶中加入co(acac)2(15.6mg,0.06mmol),4,5-双(二苯基膦)-9,9-二甲基氧杂蒽xantphos(34.8mg,0.06mmol)和thf(10ml)。将该溶液在室温搅拌10分钟。然后,在氮气下将苯基硅烷1(4.544g,42mmol)和1,5-己二烯(1.574g,19mmol)加入到瓶中。将反应瓶于室温下搅拌12小时。然后,将所得溶液真空浓缩。将粗产物用石油醚溶解,并将溶液通过硅胶短柱快速过滤。收集滤液并在减压下浓缩。剩余物通过硅胶柱层析法(石油醚)纯化,得到无色油状液体。为了改善单体的聚合性能,必须进行脱水过程以降低单体的水分含量。将cah2(500mg)加入到单体3a中,并将该混合物搅拌过夜。减压蒸馏混合物,得到无色液体的单体3a(4.307g,76%)。
[0052]
实施例2
[0053]
单体6的合成
[0054][0055]
根据改进的文献方法(参考文献十二:c.wang,w.-j.teo,s.ge,acs catal.2017,7,855.)可以方便地制备11-(苯基硅烷基)十一烷-14,5-双(二苯基膦)-9,9-二甲基氧杂蒽-醇(6)。氮气保护下,在反应瓶中加入co(acac)2(7.7mg,0.03mmol),xantphos(17.4mg,0.03mmol)和thf(5ml)。将该溶液在室温搅拌10分钟。然后,在氮气下将苯基硅烷1(0.893g,8.3mmol)和10-十一碳烯酸甲酯4(1.486g,7.5mmol)加入到瓶中。将反应瓶于室温下搅拌24小时。然后,将所得溶液真空浓缩。将粗产物用石油醚溶解,并将溶液通过硅胶短柱快速过滤。收集滤液并在减压下浓缩。向粗产物中加入et2o(30ml)。将混合物在冰浴上冷却,并小心地加入lialh4(300mg,7.5mmol)。使反应混合物升至室温,并继续搅拌12小时。通过添加酒石酸钾钠的水溶液(3g,10ml)淬灭反应。将混合物剧烈搅拌。然后将水层用乙酸乙酯(50ml
×
2)萃取。合并的有机层经硫酸钠干燥,过滤并减压浓缩。将残余物通过硅胶柱层析
法(石油醚/乙酸乙酯=50:1-10:1)纯化,得到无色液体。为了改善单体的聚合性能,必须进行脱水过程以降低单体的水分含量。向单体6中加入甲苯(20ml)和乙醇(5ml)。通过分馏蒸发共沸溶剂,得到无色液体状的无水单体6(1.881g,90%)。
[0056]
实施例3-18
[0057]
脱氢偶联聚合条件的优化
[0058]
手套箱中,在反应瓶中投入co(acac)2(底物用量的1mol%)和双膦配体(底物用量的1mol%),加入四氢呋喃(1.0ml),室温搅拌5min后,真空下抽干四氢呋喃。在反应瓶中加入硅烷3a(0.4mmol),二醇7a(0.4mmol)和反应溶剂(2.0ml)。60
–
80℃下反应24小时;然后加入2毫升二氯甲烷溶解产物,滴加15毫升冷甲醇使产物析出,去除上层溶剂,抽干得聚合产物,反应式和配体结构如下:
[0059][0060]
聚合物的数均分子量(m
n
)和分子量分布(pdi)通过凝胶色谱仪(gpc)测定,产率为分离收率,改变反应温度和溶剂种类以及催化剂和底物的比例,制备得到分子量不同的产物,详见表1。
[0061]
表1.钴催化选择性脱氢偶联聚合条件优化
a
[0062]
[0063]
实施例18-25
[0064]
aa和bb型单体选择性脱氢偶联合成聚硅醚8a-h
[0065]
手套箱中,在反应瓶中投入co(acac)2(底物用量的1mol%)和1,5-双(二苯基膦)戊烷dpppe(底物用量的1mol%),加入四氢呋喃(1.0ml),室温搅拌5min后,真空下抽干四氢呋喃。在反应瓶中加入硅烷3(0.4mmol),二醇7(0.4mmol)和正己烷(2.0ml)。60℃下反应24小时;然后加入2毫升二氯甲烷溶解产物,滴加15毫升冷甲醇使产物析出,去除上层溶剂,抽干得聚合产物,反应式和配体结构如下:
[0066][0067]
聚合物的数均分子量(m
n
)和分子量分布(pdi)通过凝胶色谱仪(gpc)测定,产率为分离收率,详见表2。
[0068]
表2.aa和bb型羟基硅烷单体脱氢偶联聚合
a
[0069][0070]
实施例26
[0071]
ab型单体脱氢偶联合成聚硅醚8i
[0072]
手套箱中,在反应瓶中投入co(acac)2(底物用量的1mol%)和dpppe(底物用量的1mol%),加入四氢呋喃(1.0ml),室温搅拌5min后,真空下抽干四氢呋喃。在反应瓶中加入单体6(0.4mmol),和正己烷(2.0ml)。60℃下反应24小时;然后加入2毫升二氯甲烷溶解产物,滴加15毫升冷甲醇使产物析出,去除上层溶剂,抽干得聚合产物,反应式和配体结构如下:
[0073][0074]
聚合物的数均分子量(m
n
)和分子量分布(pdi)通过凝胶色谱仪(gpc)测定,产率为分离收率。
[0075]
实施例27
[0076]
两步一锅法合成聚硅醚8a
[0077]
考虑到co(acac)2/xantphos催化体系能实现烯烃的区域选择性硅氢化(参考文献十一:c.wang,w.-j.teo,s.ge,acs catal.2017,7,855.),我们尝试了钴催化两步一锅法。在第一步中,将1,5-己二烯1和苯基硅烷2用作起始原料,通过硅氢化反应得到硅烷3a。然后,加入二醇7a,再通过钴催化的脱氢偶联聚合得到的聚硅醚8a,具体操作步骤如下:
[0078]
氮气保护下,向装有磁力搅拌棒的烤箱干燥的25mlschlenk瓶中装入co(acac)2(2.6mg,0.01mmol),xantphos(5.8mg,0.01mmol)和thf(1.0ml)。将该溶液在室温搅拌5分钟。然后,在氮气下将苯基硅烷(108mg,1mmol)和1,5-己二烯(41mg,0.5mmol)加入到瓶中。将反应瓶于室温搅拌5小时。然后,加入二醇7a(87mg,0.5mmol),升温至60℃搅拌24小时。反应结束后,将反应混合物冷却至室温,并通过沉淀法纯化产物。获得了聚硅醚8a(133mg,收率=57%,m
n
=6100,pdi=5.05)。
[0079][0080]
聚合物的数均分子量(m
n
)和分子量分布(pdi)通过凝胶色谱仪(gpc)测定,产率为分离收率。
[0081]
实施例28
[0082]
不对称脱氢偶联聚合合成聚硅醚8a
[0083]
向装有磁力搅拌棒的烘箱干燥的25mlschlenk烧瓶中装入co(acac)2(1.3mg,0.005mmol),(s)-(-)-(6,6'-二甲氧基联苯-2,2'-基)双(二苯基膦)(s)-meo-biphep(2.9mg,0.005mmol)和thf(3ml)在氮气下。将该溶液在室温搅拌5分钟。然后,在氮气下将单体3a(0.5mmol)和单体7a(0.5mmol)加入到烧瓶中。将烧瓶在氮气下(连接至氮气schlenk管线)在60℃下搅拌24小时。在反应时间的最后6小时内,每2小时用氮气置换反应过程中产生的h2。聚合反应结束后,将反应混合物冷却至室温,并通过沉淀法(二氯甲烷/甲醇=1/10)纯化反应物。得到具有高分子量的聚合物8a(189mg,产率=81%,m
n
=9900,pdi=1.90)。
[0084]
为了评估聚合物的立体选择性,使用甲基碘化镁在乙醚中的溶液与聚硅醚8a反应。断裂反应的步骤:在氮气下,向装有磁力搅拌棒的烘箱干燥的50ml schlenk烧瓶中加入聚硅醚8a(132mg)和乙醚(3ml)。在2分钟内滴加memgi的乙醚溶液(3m,0.4ml)。将烧瓶在氮气下于室温搅拌2小时。向烧瓶中加入己烷(20ml)。通过短硅胶柱过滤纯化产物。获得手性硅烷9(90mg,99%产率)为无色液体。
[0085][0086]
聚合物的数均分子量(m
n
)和分子量分布(pdi)通过凝胶色谱仪(gpc)测定,产率为分离收率。
[0087]
实施例29-30
[0088]
热分析
[0089]
聚硅醚的热稳定性,如质量分解5%时的温度(t5)和分解50%时的温度(t
50
)通过同步热分析仪(tga)测定,玻璃化转变温度(t
g
)通过差示扫描量热仪(dsc)测定,详见表3。
[0090]
表3.聚硅醚的热分析
a
[0091][0092][0093]
1,6-bis(phenylsilyl)hexane(3a):4.307g,76%,无色液体.1h nmr(400mhz,cdcl3)δ7.74-7.54(m,4h),7.51-7.31(m,6h),4.32(t,j=3.7hz,4h),1.52-1.37(m,8h),0.99-0.92(m,4h).
13
c nmr(100mhz,cdcl3)δ135.4,132.9,129.7,128.1,32.6,25.1,10.2.hrms calculated for c
18
h
30
nsi2[m+nh4]
+
316.1911;found 316.1911.
[0094]
11-(phenylsilyl)undecan-1-ol(6):1.881g,90%,无色液体,新化合物.1h nmr(400mhz,cdcl3)δ7.66-7.51(m,2h),7.48-7.29(m,3h),4.29(t,j=3.7hz,2h),3.64(t,j=6.7hz,2h),1.63-1.50(m,2h),1.49-1.43(m,2h),1.40-1.22(m,14h),0.99-0.91(m,2h).
13
c nmr(100mhz,cdcl3)δ135.4,133.0,129.6,128.1,63.2,33.0,33.0,29.8,29.7,29.7,29.6,29.4,25.9,25.2,10.2.hrms calculated for c
17
h
34
nosi[m+nh4]
+
296.2404;found 296.2403.
[0095]
polysilylether(8a):166mg,88%yield,无色软固体.1h nmr(400mhz,cdcl3)δ7.73-7.48(m,4h),7.47-7.28(m,6h),5.05-4.74(m,2h),3.79-3.38(m,4h),1.59-1.20(m,24h),1.01-0.80(m,4h).
13
c nmr(100mhz,cdcl3)δ135.5,134.2,130.1,128.1,65.1,32.8,32.6,29.7,29.5,25.9,23.0,14.2.
[0096]
polysilylether(8b):61mg,32%yield,无色油状物.1h nmr(400mhz,cdcl3)δ7.71-7.54(m,4h),7.52-7.28(m,6h),5.05-4.83(m,2h),3.84-3.53(m,4h),1.64-1.49(m,4h),1.47-1.17(m,22h),0.99-0.81(m,4h).
13
c nmr(100mhz,cdcl3)δ135.5,134.2,130.1,128.1,65.1,32.8,32.6,29.8,29.7,29.6,25.9,23.0,14.2.
[0097]
polysilylether(8c):177mg,89%yield,无色软固体.1h nmr(400mhz,cdcl3)δ7.82-7.49(m,4h),7.49-7.26(m,6h),5.08-4.78(m,2h),3.92-3.33(m,4h),1.65-1.10(m,
28h),1.02-0.80(m,4h).
13
c nmr(100mhz,cdcl3)δ135.5,134.2,130.1,128.1,65.1,32.8,32.6,29.8,29.6,25.9,23.0,14.2.
[0098]
polysilylether(8d):137mg,75%yield.无色软固体.1h nmr(400mhz,cdcl3)δ7.70-7.47(m,4h),7.47-7.26(m,6h),5.02-4.80(m,2h),3.81-3.47(m,4h),1.66-1.14(m,22h),1.00-0.80(m,4h).
13
c nmr(100mhz,cdcl3)δ135.5,134.2,130.1,128.1,65.0,32.8,32.6,29.7,29.5,25.9,23.0,14.2.
[0099]
polysilylether(8e):145mg,82%yield,无色油状物.1h nmr(400mhz,cdcl3)δ7.68-7.50(m,4h),7.49-7.27(m,6h),5.04-4.79(m,2h),3.79-3.49(m,4h),1.66-1.16(m,20h),1.00-0.74(m,4h).
13
c nmr(100mhz,cdcl3)δ135.5,134.2,130.1,128.1,65.0,32.8,32.5,29.5,25.8,23.0,14.2.
[0100]
polysilylether(8f):142mg,82%yield,淡黄色软固体.1h nmr(400mhz,cdcl3)δ7.78-7.29(m,10h),5.18-4.72(m,2h),3.94-3.38(m,2h),2.06-1.66(m,4h),1.57-1.15(m,12h),0.98-0.73(m,4h).
13
c nmr(100mhz,cdcl3)δ136.0,134.1,130.1,128.0,72.1,70.4,32.8,32.5,32.4,30.7,30.5,23.0,14.6.
[0101]
polysilylether(8g):158mg,85%yield,淡黄色软固体.1h nmr(400mhz,cdcl3)δ7.91-7.54(m,8h),7.53-7.28(m,6h),5.55-5.27(m,2h),3.93-3.55(m,4h),1.69-1.46(m,4h),1.40-1.06(m,12h).
13
c nmr(100mhz,cdcl3)δ136.6,134.8,134.2,134.1,130.5,128.2,65.2,32.5,29.7,29.5,25.9.
[0102]
polysilylether(8h):93mg,70%yield,无色软固体.1h nmr(400mhz,cdcl3)δ7.80-7.46(m,4h),5.12-4.86(m,2h),3.65(t,j=6.6hz,4h),1.63-1.51(m,4h),1.37-1.19(m,12h),0.51-0.39(m,6h).
13
c nmr(100mhz,cdcl3)δ138.1,133.4,64.9,32.6,29.7,29.5,25.9,-2.6.
[0103]
polysilylether(8i):88mg,80%yield.无色油状物.1h nmr(400mhz,cdcl3)δ7.67-7.54(m,2h),7.49-7.30(m,3h),5.04-4.80(m,1h),3.76-3.58(m,2h),1.60-1.50(m,2h),1.46-1.12(m,16h),0.98-0.81(m,2h).
13
c nmr(100mhz,cdcl3)δ135.6,134.3,134.2,130.1,128.1,65.1,33.3,32.6,29.8,29.7,29.6,29.5,25.9,23.1,14.3.
[0104]
(+)-1,6-bis(methyl(phenyl)silyl)hexane(9):90mg,99%yield.无色液体.[α]
20d
=+3.92(c1.30,
n
hexane),新化合物.1h nmr(400mhz,cdcl3)δ7.61-7.50(m,4h),7.44-7.30(m,6h),4.39-4.31(m,2h),1.44-1.30(m,8h),0.90-0.76(m,4h),0.34(d,j=3.8hz,6h).
13
c nmr(100mhz,cdcl3)δ136.9,134.5,129.3,128.0,33.0,24.4,13.5,-5.5.hrms calculated for c
20
h
34
nsi2[m+nh4]
+
344.2224;found344.2243.hplc:chiracel od-h column,230nm,30℃,
n
hexane/i-proh=100/0,flow=0.6ml/min,retention time 13.2min,15.1min(meso)and 17.7min(maj).
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1