一种HFPO直接生产双邻二甲苯基六氟丙酮的设备及工艺的制作方法
【技术领域】
本发明涉及一种hfpo直接生产双邻二甲苯基六氟丙酮的设备及工艺。
背景技术:
聚酰亚胺(pi)是一类重要的高分子材料,其分子链中刚性的酰亚胺结构使其具有优良的耐高温性能、环境稳定性以及卓越的机械性能等等,因此广泛应用在航空航天、机械、电工以及电子等高科技领域。但是,pi具有难熔难溶的缺点,不易成型加工,这在一定程度上限制了其应用。在pi中引入含氟基团不仅可以大大改善其溶解性,还会赋予pi更优异的物理化学、光学、电及气体分离等功能性,因此含氟聚酰亚胺具有独特优势和广阔开发前景。
用2,2-双(3,4-二羧酸)六氟丙烷二酐(六氟二酐,6fda)单体合成的含氟聚酰亚胺,其性能上主要有以下优势:1)低介电常数,普通的聚酰亚胺的介电常数在3.4左右,而加入6fda后,其介电常数可降低至2.5。2)透明性及可控的折射率,普通聚酰亚胺略带微黄色或棕色,加入6fda可使其变为透明,并且其折射率随6fda加入量而变化,因此可调。3)因为氟原子的强负电性,含氟聚酰亚胺具有更好的化学及热稳定性,其制品可以在特殊的化学环境或温度下使用。4)因为氟原子的低极性,所以含氟类聚酰亚胺具有很低的表面自由能,因此具有表面光滑、摩擦力小的特点。迄今为止,基于6fda的含氟聚酰亚胺已经在包括太空薄膜衍射光学成像系统、柔性电路板和柔性触摸屏等高科技领域获得重要应用。
2,2-二(3,4-二甲基苯基)六氟丙烷(双邻二甲苯基六氟丙烷,6fxy或boxaf)是合成6fda的原料,关于其制备方法,国内外已经有专利文献报道。
早在1967年,美国专利us3310573a就公开了二芳基六氟丙烷类化合物的合成方法,在氟化氢存在下,二摩尔芳香族化合物(如苯胺、乙酰苯胺、硝基苯胺、溴代苯胺以及甲苯、二甲苯,等等)和六氟丙酮缩合而成。2003年,朱仕正等(journaloffluorinechemistry123(2003)221-225)详细描述了双邻二甲苯基六氟丙酮的合成,在氟化氢存在下,二摩尔邻二甲苯和六氟丙酮缩合而成。作者还探索了由六氟环氧丙烷丙烷直接合成双二甲苯基六氟丙酮的可能性,发现在lewis催化剂如alcl3等的存在下,六氟环氧丙烷和邻二甲苯反应会有少量反应产物生成,但是产物中有cl杂质;而在没有上述催化剂的情况下,几乎不能得到反应产物。可以看出这种直接由hfpo/邻二甲苯一锅煮的办法并不成功。
接下来的专利技术文献主要是针对早期公开的技术方案进行优化。比如,申请公布号cn106699504a的中国专利文献提供了一种双邻二甲苯基六氟丙酮制备方法,该方法包括以下步骤:1)通过六氟丙酮三水合物与邻二甲苯在摩尔配比为1:2~1:6,加热温度为90℃~100℃加热回流条件下进行共沸脱水得到六氟丙酮一水合物;2)经共沸脱水反应后得到的含六氟丙酮一水合物的反应液可以无需提纯,直接作为原料与邻二甲苯在摩尔配比为1:0.4~2,反应温度优选为90~130℃,并在两者总重的45~80%wt的氟化氢的作用下进行液相反应得到2,2-双(3,4-二甲苯基)六氟丙烷。本发明提供的方法原料毒性低、反应压力低、易操作、反应选择性和收率高、适合工业化生产。
又如,授权公告号cn1049420c的中国专利公开了直接使用三氟甲基磺酸催化剂,以苯或苯的衍生物为原料,与六氟丙酮进行缩合反应合成双邻二甲苯基六氟丙烷的方法。类似的,申请公布号cn108395363a的中国专利文献进一步提供了以磺酸为催化剂合成双邻二甲苯基六氟丙烷的方法,包括以下步骤:1)将邻二甲苯、磺酸和溶媒按比例混合其中邻二甲苯与磺酸的摩尔比为1.0:0.1~1.0:1.0,得到混合物。2)六氟丙酮气体通入到步骤1)中的混合物,在反应温度和反应压力下,反应得到粗产物,将粗产物碱洗、水洗、浓缩后,经结晶溶媒结晶、真空干燥得到2,2-双(3,4-二甲苯基)六氟丙烷。所述磺酸选自甲基磺酸、三氟甲磺酸、1-三氟乙基磺酸、五氟乙基磺酸、丙基磺酸、1-三氟丙基磺酸、苯磺酸、4-甲基苯磺酸、4-三氟甲基苯磺酸中的至少一种。
授权公告号cn104496763b的中国专利提供了一种直接使用hfpo/邻二甲苯一锅法合成boxaf的方法,其特征是将芳香烃与无水氟化氢先在反应器中混合,再加入催化剂,然后加入六氟环氧丙烷,之后进行搅拌和加热。反应结束后,将氟化氢除去,所余产物进行精制,得到二芳基六氟丙烷化合物产品。其中,所述催化剂为sbcl5、ticl4或二者的混合物;所述芳香烃、六氟环氧丙烷、无水氟化氢按质量比1:(0.5~4):(0.5~4)混合;所述反应的温度为50到200℃,反应时间为1~12小时。本发明的方法使六氟环氧丙烷异构化和与芳香烃的缩合反应合并到一个工艺阶段中实现,减少了合成的工艺步骤,减少了生产成本和副产物生成,且大大的提高了产物的收率。类似的,专利文献公开号cn101851147a、cn104370669b和cn104326882b提供了一锅法合成boxaf的方法。但是这些专利没有披露如何克服在lewis酸催化剂存在的情况下,产物中存在cl杂质的问题。
最后,授权公告号cn101696199b的中国专利提供了另外一种双邻二甲苯基六氟丙酮的制备方法,由邻二甲苯与2,2-二氯六氟丙烷在离子液体(1-丁基-3-甲基咪唑啉四氟硼酸盐、1-丁基-3-甲基咪唑啉六氟磷酸盐,等等)中,经lewis酸(alcl3,zncl2)催化下经加热发生烷基化反应,获得4,4′-(六氟亚异丙烯基)二邻二甲苯。但是这种方法涉及的原料2,2-二氯六氟丙烷和离子液体难以得到且价格昂贵,并不适合于工业化生产。
综上所述,目前公开的boxaf制备方法有三种:1)hfa法,该方法以hfa为原料,在催化剂存在下hfa与邻二甲苯进行缩合反应得到产物。由于hfa毒性大,存在较大的运输、储藏和使用风险。不仅如此,hfa原料不容易获得且价格较高,因此生产成本比较高。2)hfpo/邻二甲苯一锅法,此方法以hfpo为原料,将hfpo异构化(生成hfa)和hfa与芳香烃的缩合反应合并到一个工艺中。尽管该方法避免了hfa的运输和储存的相关风险,但是生产过程中涉及lewis酸催化剂使用,反应中不可避免的一个问题是会产生大量的含氯副产物,难以分离。而且过程中几个反应同时进行,反应复杂程度增加,控制起来更加困难,因此会增加hfpo单耗,使生产成本升高,而且催化剂不容易回收,其排放容易造成环境污染。3)2,2-二氯六氟丙烷法。这种方法研究较少,一个很重要的问题是涉及的原料2,2-二氯六氟丙烷、离子液体难以得到且价格昂贵,并不适合于工业化生产。
技术实现要素:
本发明要解决的技术问题,在于提供一种hfpo直接生产双邻二甲苯基六氟丙酮的设备及工艺,所用原料毒性低,安全环保,而且成本更加低;缩合反应产物中不含cl杂质,容易分离;反应催化剂可直接回收利用,既节省了成本又避免对环境产生危害;可以实现hfa生产和消耗同步进行,避免了hfa运输和储存带来的风险。
本发明是这样实现的:
一种hfpo直接生产双邻二甲苯基六氟丙酮的设备,所述设备包括异构釜、气液分离釜、精馏柱、冷凝器、hfa储罐、缩合釜,所述异构釜上设置有hfpo进料口和异构反应催化剂进料口,所述异构釜底部的第一循环管道通过第一循环泵连接至气液分离釜的底部,所述气液分离釜的上方与所述异构釜的上方通过第二循环管道相连接,所述第二循环管道上设置有第二循环泵;
所述气液分离釜的顶部通过管道连接至精馏柱底部,所述精馏柱的上端分别连接至异构釜的hfpo进料口和hfa储罐,且所述精馏柱和hfa储罐之间的连接管道上还设置有冷凝器;
所述hfa储罐连接至所述缩合釜,且所述缩合釜上还设置有原料进料口和缩合反应催化剂进料口;所述异构釜和缩合釜内均设置有搅拌器。
进一步地,所述设备还包括hf回收罐,所述hf回收罐与所述缩合釜的缩合反应催化剂进料口相连。
本发明还涉及一种hfpo直接生产双邻二甲苯基六氟丙酮的工艺,所述工艺步骤如下:
步骤1、将反应装置加热抽真空除水;
步骤2、用高纯氮气置换反应系统内的空气,除氧;
步骤3、开启搅拌器搅拌系统,向异构釜计量加入异构反应催化剂lewis酸;
步骤4、计量快速通入hfpo,升温反应;
步骤5、待异构釜中hfpo完全转化为hfa后,开启第一循环泵,异构釜中的反应物料进入气液分离釜中,发生气液分离;
从气液分离釜气化的hfa在精馏柱中与其中夹带的少量异构反应催化剂lewis酸进一步分离,纯度合格的hfa气体经冷凝器冷却后收集到hfa储罐中,而纯度不合格的hfa气体则重新回到异构釜中,夹带的少量异构反应催化剂lewis酸则回到气液分离釜,再经第二循环泵输送到异构釜,继续用于hfpo异构化反应催化剂;
步骤6、将hfa储罐中的hfa计量加入用于hfa缩合反应的缩合釜,计量加入缩合反应催化剂,然后再计量加入邻二甲苯;
步骤7、升温反应,待hfa完全反应,得到双邻二甲苯基六氟丙酮粗品。
进一步地,所述工艺步骤还包括:
步骤8、待缩合反应完成后,对双邻二甲苯基六氟丙酮粗品降温,蒸馏除去缩合反应催化剂,缩合反应催化剂回收到缩合反应催化剂储罐中循环利用;
步骤9、经步骤8后的残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠溶液、去离子水萃取,除去水溶性杂质;再用有机溶剂萃取、浓缩干燥重结晶得到最终产品双邻二甲苯基六氟丙酮。
进一步地,所述异构反应催化剂lewis酸为sbf5、al2o3、tio2、wo2、alcl3、albr3、sncl4、fecl3、cucl2或zrocl2;
所述异构反应催化剂lewis酸的用量为hfpo加入量的5-20%,所述异构反应的温度为30~150℃。
进一步地,所述lewis酸为sbf5;所述异构反应催化剂lewis酸的用量为hfpo加入量的10%,所述异构反应的温度为90℃。
进一步地,所述缩合反应催化剂为磺酸和hf;
所述hf用量为六氟丙酮hfa与邻二甲苯总重的40~80%wt;
所述hfa与邻二甲苯的摩尔配比为1:2~1:6;
所述hfa缩合反应温度为60~180℃。
进一步地,所述hfa缩合反应溶剂和催化剂为hf;
所述hfa缩合反应温度是90~150℃。
进一步地,所述hfpo异构化反应压力为1~20atm;所述hfpo异构化反应时间为1~24小时;
所述hfa缩合反应压力为1~20atm;所述hfa缩合反应时间为1~24小时。
进一步地,步骤8中,所述hfa缩合反应完成后,缩合反应催化剂hf回收温度为60~180℃。
本发明具有如下优点:
本发明所述hfpo异构化反应过程是在一个外循环反应器内进行,包括一个具有搅拌器的异构釜和与之连接的气液分离釜组成。hfpo异构化反应在异构釜内进行,反应产物与催化剂的分离则在气液分离釜罐中完成。所述气液分离釜进一步与精馏柱连接,精馏柱与冷凝器连接,冷凝器与hfa储罐连接。反应过程中,物料在异构釜和气液分离釜中循环流动,反应和分离过程同时进行。通过这样的一个反应分离系统,可以实现hfpo的异构化反应、产物分离纯化和储存;以及可以实现hfa生产(hfpo异构化)和消耗(hfa缩合)同步进行,因此最大限度地降低了hfa运输和储存带来的风险。
并且,相较于hfa法,本发明所用原料为hfpo毒性低,储存、运输和使用均较为安全环保,而且成本更加低;相较于hfpo一锅法,本发明hfa和邻二甲苯缩合过程中使用hf作为溶剂和催化剂,不使用lewis酸,因此反应产物中不含cl杂质,容易分离。生产过程中溶剂hf以及催化剂sbf5可以直接回收利用,既节省了成本又避免对环境产生危害。
【附图说明】
下面参照附图结合实施例对本发明作进一步的说明。
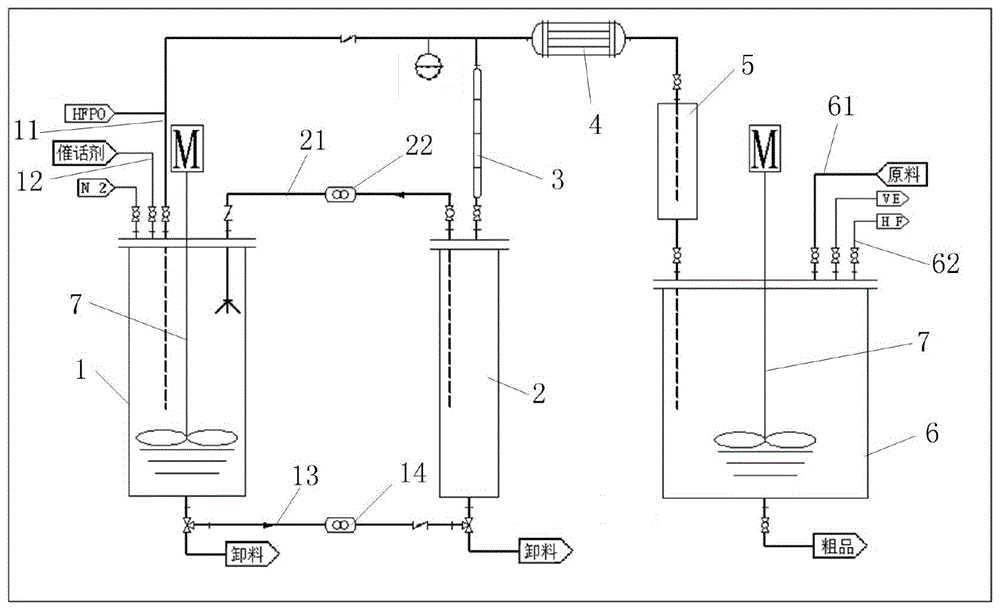
图1为本发明一种hfpo直接生产双邻二甲苯基六氟丙酮的设备示意图。
【具体实施方式】
参阅图1所示,本发明涉及一种hfpo直接生产双邻二甲苯基六氟丙酮的设备,所述设备包括异构釜1、气液分离釜2、精馏柱3、冷凝器4、hfa储罐5、缩合釜6,所述异构釜1上设置有hfpo进料口11和异构反应催化剂进料口12,所述异构釜1底部的第一循环管道13通过第一循环泵14连接至气液分离釜2的底部,所述气液分离釜2的上方与所述异构釜1的上方通过第二循环管道21相连接,所述第二循环管道21上设置有第二循环泵22;位于所述气液分离釜2处的所述第二循环管道21一端伸入至所述气液分离釜2底部。
所述气液分离釜2的顶部通过管道连接至精馏柱3底部,所述精馏柱3的上端分别连接至异构釜1的hfpo进料口11和hfa储罐5,且所述精馏柱3和hfa储罐5之间的连接管道上还设置有冷凝器4;
所述hfa储罐5连接至所述缩合釜6,且所述缩合釜6上还设置有原料进料口61和缩合反应催化剂进料口62;所述异构釜1和缩合釜6内均设置有搅拌器7。
所述设备还包括hf回收罐,所述hf回收罐与所述缩合釜6的缩合反应催化剂进料口62相连。
本发明还涉及在上述反应装置中实施的一种hfpo直接生产双邻二甲苯基六氟丙酮的工艺,所述工艺步骤如下:
步骤1、将反应装置加热抽真空除水;
步骤2、用高纯氮气置换反应系统内的空气,除氧;
步骤3、开启搅拌器搅拌系统,室温下,向异构釜计量加入异构反应催化剂lewis酸;所述异构反应催化剂lewis酸为sbf5、al2o3、tio2、wo2、alcl3、albr3、sncl4、fecl3、cucl2或zrocl2;较优的,所述lewis酸为sbf5;
所述异构反应催化剂lewis酸的用量为hfpo加入量的5-20%;较优的,所述异构反应催化剂lewis酸的用量为hfpo加入量的10%,
步骤4、计量快速通入hfpo,升温反应;所述异构反应的温度为30~150℃;较优的,所述异构反应的温度为90℃。温度太高异构反应容易出现副产物,温度太低异构化反应速度太慢。
步骤5、待异构釜中hfpo完全转化为hfa后,开启第一循环泵,异构釜中的反应物料进入气液分离釜中,发生气液分离;
从气液分离釜气化的hfa在精馏柱中与其中夹带的少量异构反应催化剂lewis酸进一步分离,纯度合格的hfa气体经冷凝器冷却后收集到hfa储罐中,而纯度不合格的hfa气体则重新回到异构釜中,夹带的少量异构反应催化剂lewis酸则回到气液分离釜,再经第二循环泵输送到异构釜,继续用于hfpo异构化反应催化剂;
步骤6、室温下,将hfa储罐中的hfa计量加入用于hfa缩合反应的缩合釜,计量加入缩合反应催化剂,然后再计量加入邻二甲苯;所述缩合反应催化剂为磺酸和hf;所述hf用量为六氟丙酮hfa与邻二甲苯总重的40~80%wt;所述hfa与邻二甲苯的摩尔配比为1:2~1:6;较优的,所述hfa缩合反应溶剂和催化剂为hf。
步骤7、升温反应,待hfa完全反应,得到双邻二甲苯基六氟丙酮粗品。所述hfa缩合反应温度为60~180℃;较优的,所述hfa缩合反应温度是90~150℃。
步骤8、待缩合反应完成后,对双邻二甲苯基六氟丙酮粗品降温,蒸馏除去缩合反应催化剂,缩合反应催化剂回收到缩合反应催化剂储罐中循环利用;缩合反应催化剂hf回收温度为60~180℃。
步骤9、经步骤8后的残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠溶液、去离子水萃取,除去水溶性杂质;再用有机溶剂萃取、浓缩干燥重结晶得到最终产品双邻二甲苯基六氟丙酮。
所述hfpo异构化反应压力为1~20atm;所述hfpo异构化反应时间为1~24小时;
所述hfa缩合反应压力为1~20atm;所述hfa缩合反应时间为1~24小时。
以下结合实施例对本发明作进一步说明。但不得将这些实施例解释为对本发明保护范围的限制。凡属于与本发明等效的技术方案,均属于本发明的保护范围。
比较例一
反应在1000ml蒙乃尔反应系统中进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。室温下,向反应釜通入hfpo(165.37g,0.996mol),计量加入无水hf(200g,10mol),然后计量加入邻二甲苯(212.2g,2mol)。升温130℃,反应釜压力10公斤。反应16h时间后。降温,蒸馏除去溶剂hf,溶剂hf回收到储罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥。产物收率为0%。
比较例二
反应在1000ml蒙乃尔反应系统中进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。室温下,向反应釜通入hfpo(165.37g,0.996mol),计量加入无水hf(200g,10mol),催化剂alcl3(13.3g,0.1mol),然后计量加入邻二甲苯(212.2g,2mol)。升温130℃,反应釜压力10公斤。反应16h时间后。降温,蒸馏除去溶剂hf,溶剂hf回收到储罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥,再用乙醇重结晶,湿品40℃真空干燥12小时,得到产物(107.67g,0.299mol),包括含氯杂质在内的产品总收率30%。
实施例一
六氟环氧丙烷直接连续法合成2,2-二(3,4-二甲基苯基)六氟丙烷反应在一套2台1000ml蒙乃尔反应系统中模拟进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。在氮气保护下,向hfpo异构化反应釜即异构釜内通入催化剂sbf5(21.67g,0.1mol),然后快速计量通入六氟环氧丙烷(166.02g,1mol),升温到90℃,催化重排得到六氟丙酮(hfa)(165.37g,0.996mol)。降温对hfpo异构化反应产物hfa进行精馏,hfa经冷凝器冷却后进入hfa储罐中,催化剂sbf5回流到hfpo异构化反应釜中。精馏结束后,室温下hfpo异构化反应釜继续快速计量通入hfpo,并根据需要加入少量催化剂sbf5,准备进入第二次反应。
室温下,往2,2-二(3,4-二甲基苯基)六氟丙烷反应合成釜即缩合釜通入hfa储罐中hfa(165.37g,0.996mol),计量加入无水hf(200g,10mol)。然后计量加入邻二甲苯(212.2g,2mol)。升温130℃,缩合釜压力10公斤。反应16h时间后,hfa完全反应。降温,蒸馏除去溶剂hf,溶剂hf回收到hf回收罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥,再用乙醇重结晶,湿品40℃真空干燥12小时,得到产物(218g,0.605mol),gc纯度为99.8%,计算产率为60.5%。
实施例二
六氟环氧丙烷直接连续法合成2,2-二(3,4-二甲基苯基)六氟丙烷反应在一套2台1000ml蒙乃尔反应系统中模拟进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。在氮气保护下,向实施例一所述hfpo异构化反应釜即异构釜内快速计量通入六氟环氧丙烷共计(166.02g,1mol),升温到90℃,催化重排得到六氟丙酮(hfa)(164.53g,0.991mol)。降温对hfpo异构化反应反应产物hfa进行精馏,hfa经冷凝器冷却后进入hfa储罐中,催化剂sbf5回流到hfpo异构化反应釜中。精馏结束后,继续往hfpo异构化反应釜中快速计量通入hfpo,并根据需要加入少量催化剂sbf5,准备进入第二次反应。
室温下,往2,2-二(3,4-二甲基苯基)六氟丙烷反应合成釜即缩合釜中通入hfa储罐中的hfa164.53g(0.991mol),计量加入无水hf(200g,10mol)。然后计量加入邻二甲苯(212.2g,2mol)。升温130℃,缩合釜压力10公斤。反应16h时间后,hfa完全反应。降温,蒸馏除去溶剂hf,溶剂hf回收到hf回收罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥,然后用乙醇重结晶,湿品40℃真空干燥12小时,得到产物(210.8g,0..585mol),gc纯度为99.6%,计算产率为58.5%。
实施例三
六氟环氧丙烷直接连续法合成2,2-二(3,4-二甲基苯基)六氟丙烷反应在一套2台1000ml蒙乃尔反应系统中模拟进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。在氮气保护下,向hfpo异构化反应釜即异构釜内通入催化剂sbf5(21.67g,0.1mol),然后快速计量通入六氟环氧丙烷(166.02g,mol),升温到90℃,催化重排得到六氟丙酮(hfa)(165.37g,0.996mol)。降温对hfpo异构化反应产物hfa进行精馏,hfa经冷凝器冷却后进入hfa储罐中,催化剂sbf5回流到hfpo异构化反应釜中。精馏结束后,室温下继续往hfpo异构化反应釜快速计量通入hfpo,并根据需要加入少量催化剂sbf5,准备进入第二次反应。
室温下,往2,2-二(3,4-二甲基苯基)六氟丙烷反应合成釜即缩合釜通入hfa储罐中的hfa(165.37g,0.996mol),计量加入无水hf(200g,10mol)。然后计量加入邻二甲苯(212.2g,2mol)。升温150℃,缩合釜压力11公斤。反应16h时间后,hfa完全反应。降温,蒸馏除去溶剂hf,溶剂hf回收到hf回收罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥,再用乙醇重结晶,湿品40℃真空干燥12小时,得到产物(246.8g,0.685mol),gc纯度为99.8%,计算产率为68.5%。
实施例四
六氟环氧丙烷直接连续法合成2,2-二(3,4-二甲基苯基)六氟丙烷反应在一套2台1000ml蒙乃尔反应系统中模拟进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。在氮气保护下,向hfpo异构化反应釜即异构釜内通入催化剂sbf5(21.67g,0.1mol),然后快速计量通入六氟环氧丙烷(166.02g,mol),升温到60℃,催化重排得到六氟丙酮(hfa)(165.37g,0.996mol)。降温对hfpo异构化反应产物hfa进行精馏,hfa经冷凝器冷却后进入hfa储罐中,催化剂sbf5回流到hfpo异构化反应釜中。精馏结束后,室温下往hfpo异构化反应釜继续快速计量通入hfpo,并根据需要加入少量催化剂sbf5,准备进入第二次反应。
室温下,往2,2-二(3,4-二甲基苯基)六氟丙烷反应合成釜即缩合釜通入hfa储罐中的hfa(165.37g,0.996mol),计量加入无水hf(200g,10mol)。然后计量加入邻二甲苯(212.2g,2mol)。升温130℃,缩合釜压力10公斤。反应16h时间后,hfa完全反应。降温,蒸馏除去溶剂hf,溶剂hf回收到hf回收罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥,再用乙醇重结晶,湿品40℃真空干燥12小时,得到产物(241.4g,0.67mol),gc纯度为99.8%,计算产率为67.0%。
实施例五
六氟环氧丙烷直接连续法合成2,2-二(3,4-二甲基苯基)六氟丙烷反应在一套2台1000ml蒙乃尔反应系统中模拟进行。反应系统加热抽真空除水,用高纯氮气吹扫整个系统除去其中的氧气。在氮气保护下,向hfpo异构化反应釜即异构釜内通入催化剂sbf5(21.67g,0.1mol),然后快速计量通入六氟环氧丙烷(166.02g,mol),升温到60℃,催化重排得到六氟丙酮(hfa)(165.37g,0.996mol)。降温对hfpo异构化反应产物hfa进行精馏,hfa经冷凝器冷却后进入hfa储罐中,催化剂sbf5回流到hfpo异构化反应釜中。精馏结束后,室温下往hfpo异构化反应釜继续快速计量通入hfpo,并根据需要加入少量催化剂sbf5,准备进入第二次反应。
室温下,往2,2-二(3,4-二甲基苯基)六氟丙烷反应合成釜即缩合釜中通入hfa储罐中的hfa(165.37g,0.996mol),计量加入无水hf(200g,10mol)。然后计量加入邻二甲苯(212.2g,2mol)。升温130℃,缩合釜压力10公斤。反应16h时间后,hfa完全反应。降温,蒸馏除去溶剂hf,溶剂hf回收到hf回收罐中循环利用。残留物用泵输送到超离心萃取塔中,先后用10%的氢氧化钠(naoh)溶液、去离子水萃取到中性,浓缩、干燥,再用乙醇重结晶,湿品40℃真空干燥12小时,得到产物(250.4g,0.695mol),gc纯度为99.8%,计算产率为69.5%。
综上可知,相比于现有技术,本发明具有明显优势。首先,本发明所用原料为hfpo,其毒性低,储存、运输和使用均较hfa法更加安全环保,而且原料供应稳定,成本和hfa相比更低。hfpo异构化反应和hfa缩合反应分别是两个串联的反应釜中进行,过程中hfa生产出来后立刻被消耗,减少了hfa运输和储存带来的风险。其次,相较于现有hfpo一锅法,hfa和邻二甲苯缩合过程中使用hf作为溶剂和催化剂,不使用lewis酸,缩合过程中不会生成cl杂质,产物容易分离。还可以减少hfa缩合过程中副反应的发生,降低hfpo单耗。反应结束后,溶剂和催化剂hf容易回收再利用。最后,hfpo异构化过程中催化剂五氟化锑(sbf5)和hfa在气液分离罐中分离,实现催化剂sbf5直接回收利用,既减少排放,又降低成本。
虽然以上描述了本发明的具体实施方式,但是熟悉本技术领域的技术人员应当理解,我们所描述的具体的实施例只是说明性的,而不是用于对本发明的范围的限定,熟悉本领域的技术人员在依照本发明的精神所作的等效的修饰以及变化,都应当涵盖在本发明的权利要求所保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!