一种三/二氟乙醇化试剂及其制备方法与应用与流程
1.本发明属于有机合成技术领域,涉及一种三/二氟乙醇化试剂及其制备方法与 应用。
背景技术:
2.化合物的氟原子引入已经成为药物等合成中常用的策略,氟原子的引入能提高 或改变有机化合物的化学,物理和生物性质[a)p.kirsch,modern fluoroorganicchemistry:synthesis,reactivity,applications,wiley
‑
vch,2013;b)j.
‑
p.b
é
gu
é
,d. bonnet
‑
delpon,bioorganic and medicinal chemistry wiley
‑
vch,weinheim,2008; c)c.ni,m.hu,j.hu,chem.rev.2015,115,765;d)liang,t.;neumann,c.n.; ritter,t.angew.chem.,int.ed.2013,52,8214;angew.chem.2013,125,8372.]。常 用的氟烷基硅试剂例如rupert
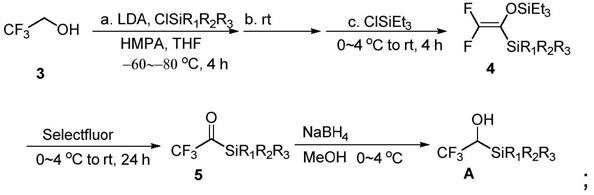
‑
prakash试剂和tmscf2h等在有机氟化合物合成中 被广泛应用,但这类经典地用阴离子活化c
‑
si键形成碳负离子中间体与醛类化合 物亲核加成得到目标产物的方法仍然有一些不足之处:1)含有多醛基的化合物中 的区域选择性难以控制;2)许多醛类化合物尤其是烷基醛不稳定,特定的醛需要 多步合成[a)y.he,m.
‑
m.tian,x.
‑
y.zhang,x.
‑
s.fan,asian j.org.chem.2016, 5,1318;b)w.ou,g.zhang,j.wu,c.su,acs cat.2019,9,5178
‑
5183.]。
[0003]
为了解决以上问题,本申请人研发了两种试剂,分别为三氟乙醇化试剂(a) 和二氟乙醇化试剂(b),这两种试剂化学性质稳定,试剂参与反应兼容性好,可 应用于药物分子或者其他功能分子的后程官能团化反应中,并且利用自由基取代/ 加成的方法实现了三/二氟乙醇α
‑
烯丙基化反应,烷基化反应,烯基化反应,得到 了一系列三/二氟甲基取代高烯丙基醇类化合物,烷基取代的三/二氟乙醇化合物和 三/二氟甲基取代烯丙醇类化合物。
技术实现要素:
[0004]
本发明所要解决的技术问题是针对现有技术中存在的上述不足,提供一种新 型的三/二氟乙醇化试剂及其制备方法与应用,该三/二氟乙醇化试剂原料易得、制 备工艺简单、条件温和、底物适用范围广,为合成含有三/二氟乙醇结构单元的药 物分子等产物提供了一种非常高效、汇聚的方法。
[0005]
为解决上述技术问题,本发明提供的技术方案是:
[0006]
一种三氟乙醇化试剂(a),其化学结构式为:
[0007][0008]
其中r1,r2,r3代表硅原子上的取代基团,r1,r2,r3各自独立选自芳基, 杂芳基,烷基。
[0009]
本申请所述芳基是指任选取代的芳香烃基,其具有6
‑
约20个,如6
‑
12个或 6
‑
10个成环碳原子,其可以是单环芳基、双环芳基或更多环芳基。双环芳基或更 多环芳基可以是
一个单环芳基与其它独立环,如脂环、杂环、芳环、芳杂环相稠合。
[0010]
本申请所述芳基可带有一个或者多个取代基,所述取代基不以任何方式限定, 常见的取代基例如芳基、烷基、酯基、氰基、硝基、酰胺基、磺酰基、烷氧基,烯 基,炔基,醛基,羟基和卤素等。所述芳香基上可带有这些取代基中的一种或多种, 当具有多个取代基时,这多个取代基可以相同或不同。
[0011]
本申请所述杂芳基是指任意取代的杂芳基,其包含约5
‑
约20个,如5
‑
12个 或5
‑
10个骨架成环原子,其中至少一个成环原子为杂原子,所述杂原子独立地选 自氧、氮、硫、磷、硅、硒和锡中的杂原子,但不限于此。所述基团的环不包含两 个相邻的o或s原子。杂芳基包括单环杂芳基(具有一个环)、双环杂芳基(具 有两个环)或多环杂芳基(具有两个以上的环)。在环中出现两个或更多杂原子的 实施方式中,所述两个或更多杂原子可彼此相同,或者所述两个或更多杂原子中的 一些或全部彼此不同。双环杂芳基或更多环杂芳基可以是一个单环杂芳基与其它独 立环,如脂环、杂环、芳环、芳杂环相稠合(可统称为稠合环杂芳基)。单环杂芳 基的非限定性实施例包括5
‑
约12个、5
‑
约10个、5
‑
约7个或6个骨架成环原子的 单环杂芳基,例如其非限定性实施例包括吡啶基;稠合环杂芳基包括苯并咪唑基、 喹啉基、吖啶基。杂芳基的其它实施例包括但不限于:吡啶、嘧啶、吡嗪、哒嗪、 三嗪、呋喃、噻吩、咪唑、三唑、四唑、噻唑、异噻唑、1,2,4
‑
噻二唑、吡咯、吡 唑、噁唑、异噁唑、噁二唑、苯并呋喃、苯并噻吩、苯并噻唑、吲哚、吲唑、喹啉、 异喹啉、嘌呤、咔唑、苯并咪唑、吡咯并吡啶、吡咯并嘧啶、吡唑并吡啶、吡唑并 嘧啶等。吖啶基、吩嗪基、苯并噁唑基、苯并噻二唑基、苯并噁二唑基、苯并三唑 基、异喹啉基、氮茚基、异噻唑基、异氮杂茚基、噁二唑基、嘌呤基、酞嗪基、蝶 啶基、喹唑啉基、喹噁啉基、三嗪基和噻二唑基等,及其氧化物,例如吡啶基
‑
n
‑ꢀ
氧化物等。
[0012]
本申请所述烷基是指任选取代的饱和脂肪族烃类,为直链结构,环状结构或 者支链结构,优选具有1
‑
约20个碳原子,例如具有1
‑
约10个碳原子,具有1
‑
约 8个碳原子,或1
‑
约6个碳原子,或1
‑
约4个碳原子或1
‑
约3个碳原子。本申请 的烷基实施例包括但不限于甲基、乙基、正丙基、异丙基、2
‑
甲基
‑1‑
丙基、2
‑
甲基
ꢀ‑
2丙基、2
‑
甲基
‑1‑
丁基、3
‑
甲基
‑1‑
丁基、2
‑
甲基
‑3‑
丁基、2,2
‑
二甲基
‑1‑
丙基、2
‑ꢀ
甲基
‑1‑
戊基、3
‑
甲基
‑1‑
戊基、4
‑
甲基
‑1‑
戊基、2
‑
甲基
‑2‑
戊基、3
‑
甲基
‑2‑
戊基、4
‑ꢀ
甲基
‑2‑
戊基、2,2
‑
二甲基
‑1‑
丁基、3,3
‑
二甲基
‑1‑
丁基、2
‑
乙基
‑1‑
丁基、正丁基、异 丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、叔戊基和己基,以及更长的烷 基基团,如庚基和辛基等。这些烷基上可以含有一个或多个取代基,这些取代基可 以但不仅限于芳基,烷基,芳酰基,烷酰基,取代氧酰基,烷氧基,卤素,烷氧基 等,烯基,炔基,杂芳基,这些取代基可以在烷基的不同位置,可以为一个,也可 以为多个,多个官能团取代时,官能团种类可以相同也可以不同,位置可以相同也 可以不相同。
[0013]
本发明还提供一种二氟乙醇化试剂(b),其化学结构式为:
[0014][0015]
其中r1,r2,r3代表硅原子上的取代基团,r1,r2,r3各自独立选自芳基, 杂芳基,烷基。
[0016]
本发明还包括上述三氟乙醇化试剂(a)的制备方法,其化学方程式为:
[0017][0018]
具体步骤为:
[0019]
1)将r1、r2、r3三取代的氯硅烷、三氟乙醇(3)、六甲磷酰三胺(hmpa) 加入溶剂(四氢呋喃)中,降温至
‑
80~
‑
60℃,用注射泵逐滴滴加二异丙基胺基锂 (lda),滴加完毕后充分搅拌,然后升温至室温,再搅拌直至三氟乙醇消耗完毕, 在0~4℃条件下加入三乙基氯硅烷,搅拌一定时间后,经柱层析分离得到化合物4;
[0020]
2)将氟化剂(优选为select
‑
fluor)加入乙腈和二氯甲烷(dcm)的混合溶 剂(乙腈与二氯甲烷体积比4:1)中,在0~4℃条件下加入化合物4,随后于室温 下反应8~12h,用水淬灭反应,经柱层析分离获得化合物5;
[0021]
3)将化合物5加入甲醇中,分批加入硼氢化钠,在0~4℃下进行反应,待反 应完毕后,加水淬灭反应,经柱层析分离得到三氟乙醇化试剂a。
[0022]
按上述方案,步骤1)所述r1、r2、r3三取代的氯硅烷与三氟乙醇及lda的物 质的量比为1:1:3.5。
[0023]
按上述方案,步骤2)所述氟化剂与化合物4物质的量比为2:1。
[0024]
按上述方案,步骤3)所述硼氢化钠总加入量与化合物5的物质的量之比为1: 1。
[0025]
本发明还包括上述二氟乙醇化试剂(b)的制备方法,其化学方程式为:
[0026][0027]
具体步骤为:
[0028]
1)将化合物4加入溶剂(thf)中,在0~4℃条件下向所得溶液中滴加浓盐 酸,滴加完毕后升温至室温搅拌反应,反应完全后,加水淬灭反应,经柱层析分离 得到化合物6;
[0029]
2)将化合物6溶于溶剂(甲醇)中,在0~4℃条件下分批加入硼氢化钠进行 反应,反应完全后,加水淬灭反应,经柱层析分离得到二氟乙醇化试剂b。
[0030]
按上述方案,步骤1)所述化合物4与浓盐酸中hcl摩尔比为1:10。
[0031]
按上述方案,步骤2)所述硼氢化钠与化合物6摩尔比为1.1:1,硼氢化钠 分三批加入。
[0032]
本发明还包括根据上述三氟乙醇化试剂(a)烯丙基化制备三氟甲基高烯丙 基醇类化合物的方法,具体方法为:在氮气保护下,三氟乙醇化试剂,烯丙基砜c, 催化剂d,氧化剂e,在50
‑
100℃的有机溶剂f中搅拌反应,反应结束经四丁基 氟化铵(tbaf)溶液淬灭反应后分离提纯,即可得到对应α
‑
三氟甲基高烯丙基醇 类化合物(g)。
[0033]
本发明方法的反应式可表示如下:
[0034][0035]
其中,r基团代表烯丙基砜上的取代基团,为芳基,杂芳基,烷基,取代烷 基(包括氧原子,氮原子基团取代的烷基),芳酰基,烷酰基,取代氧酰基,取代 胺酰基,卤素,磺酰基,取代硫酰基,烯基,炔基,氰基,硝基,胺基,醛基等;
[0036]
催化剂d优选使用锰催化剂来促进反应,可采用的锰催化剂包括二价或者三 价的锰化合物,例如:二水合醋酸锰(iii),四水合醋酸锰(ii),磷酸锰(iii), 乙酰丙酮锰(iii)等;
[0037]
氧化剂e优选过氧化物来促进反应,可采用的过氧化物包括tbhp(叔丁基 过氧化氢),dcp(过氧化二异丙苯),dtbp(二叔丁基过氧化物),tbpb(过 苯甲酸特丁酯)等;
[0038]
溶剂f是常规溶剂,选自甲醇、乙醇、异丙醇、叔丁醇、四氢呋喃、2
‑
甲基 四氢呋喃、乙醚、二甲基乙二醚、甲基叔丁基醚、1,4
‑
环氧六烷、1,3
‑
环氧六烷、 二氯甲烷、1,2
‑
二氯乙烷、氯仿、四氯化碳、c4
‑
12的饱和烷烃、c3
‑
12的氟代或 者氯代烷烃、苯、甲苯、二甲苯、三甲苯、二甲亚砜、n,n
‑
二甲基甲酰胺(dmf)、 n,n
‑
二甲基乙酰胺、丙酮、n
‑
甲基吡咯烷酮、乙腈、c3
‑
12的饱和烷基腈等。
[0039]
优选的反应条件为:在50~100℃下进行反应。
[0040]
按上述方案,所述芳酰基是指具有以下结构的官能团:
[0041][0042]
其中官能团7中r代表芳基上的取代基团,字母n代表在芳基上取代基团的 个数,0≤n≤4,所述官能团不以任何方式限定,常见的取代基芳基、烷基、酯基、 氰基、硝基、酰胺基、磺酰基、烷氧基和卤素等,当含有多个取代基团时,这些官 能团可以相同也可以不相同。官能团8中x表示构成环状芳基结构的杂原子或者 杂原子团,n,s,o等,r’代表杂环芳环上的取代基团,字母m代表取代基的个 数0≤m≤3,所述官能团不以任何方式限定,常见的取代基芳基、烷基、酯基、氰 基、硝基、酰胺基、磺酰基、烷氧基和卤素等,当含有多个取代基团时,这些官能 团可以相同也可以不相同。
[0043]
按上述方案,所述烷酰基是指具有以下结构的官能团:
[0044][0045]
其中r代表烷基。
[0046]
按上述方案,所述取代氧酰基是指具有以下结构的官能团:
[0047]
[0048]
其中r表示在氧上取代的官能团,选自烷基,芳基,天然有机化合物切块, 其中天然有机化合物切块为含有羟基的天然有机化合物,包括薄荷醇,胆固醇,睾 丸酮,薯蓣皂素,表雄甾酮,维生素e,雌二醇等。
[0049]
按上述方案,所述取代胺酰基指具有以下结构的官能团:
[0050][0051]
其中r1和r2各自独立选自芳基或烷基。这两个取代基可以相同也可以不相 同。
[0052]
本发明还包括根据上述三氟乙醇化试剂(a)与丙烯酰胺通过烷基化反应制 备三氟甲基烷基醇类化合物的方法,具体为:在氮气保护下,三氟乙醇化试剂,丙 烯酰胺h,催化剂d,氧化剂e,在50
‑
100℃的有机溶剂f中搅拌反应,反应结 束经四丁基氟化铵溶液淬灭反应后分离提纯,即可得到对应α
‑
三氟甲基烷基醇类 化合物i。
[0053]
本发明方法的反应式可表示如下:
[0054][0055]
其中r基团代表丙烯酰胺芳环上的取代基团,包括芳基,烷基,芳酰基,烷 酰基,取代氧酰基,卤素,氰基,硝基和烷氧基等。取代基可以是一个,也可以是 多个,当含有多个取代基时,这些取代基可以相同也可以不相同。r1表示氮原子 上的另一个取代基团,可以为芳基,烷基,芳酰基,烷酰基等。
[0056]
本发明还包括根据上述三氟乙醇化试剂(a)与肉桂酸通过烯基化反应制备 三氟甲基烯丙基醇类化合物的方法,具体为:在氮气保护下,三氟乙醇化试剂a, 肉桂酸j,催化剂d,氧化剂e,在50
‑
100℃的有机溶剂f中搅拌反应,反应结 束经四丁基氟化铵溶液淬灭反应后分离提纯,即可得到对应α
‑
三氟甲基烯丙基醇 类化合物k。
[0057]
本发明方法的反应式可表示如下:
[0058][0059]
其中r基团代表肉桂酸芳环上的取代基团,为芳基,烷基,芳酰基,烷酰基, 取代氧酰基,卤素,烷氧基等。这些取代基可以是一个,也可以是多个,当含有多 个取代基时,这些取代基可以相同也可以不相同。
[0060]
本发明还包括上述二氟乙醇化试剂(b)与烯丙基砜c反应制备二氟甲基高 烯丙基醇类化合物g的方法,具体为:在氮气保护下,二氟乙醇化试剂b,烯丙 基砜c,催化剂d,氧化剂e,在50
‑
100℃的有机溶剂f中搅拌反应,反应结束 经四丁基氟化铵溶液淬灭反应后分离提纯,即可得到α
‑
二氟甲基高烯丙基醇类化 合物g。
[0061]
本发明还包括上述二氟乙醇化试剂(b)与丙烯酰胺h反应制备二氟甲基烷 基醇类
化合物i的方法,具体为:在氮气保护下,二氟乙醇化试剂b,丙烯酰胺h, 催化剂d,氧化剂e,在50
‑
100℃的有机溶剂f中搅拌反应,反应结束经四丁基 氟化铵溶液淬灭反应后分离提纯,即可得到α
‑
二氟甲基烷基醇类化合物i。
[0062]
本发明还包括上述二氟乙醇化试剂(b)与肉桂酸j反应制备二氟甲基烯丙 基醇类化合物k的方法,具体为:在氮气保护下,二氟乙醇化试剂b,肉桂酸j, 催化剂d,氧化剂e,在50
‑
100℃的有机溶剂f中搅拌反应,反应结束经四丁基 氟化铵溶液淬灭反应后分离提纯,即可得到α
‑
二氟甲基烯丙基醇类化合物k。
[0063]
上述方法的反应式可表示如下:
[0064][0065]
其中,r2,r3,r4基团分别代表烯丙基砜,肉桂酸和丙烯酰胺化合物上的取 代基团,包括芳基,烷基,芳酰基,烷酰基,取代氧酰基,烷氧基,卤素,烷氧基 等,烯基,炔基,杂芳基,取代硫酰基。
[0066]
本发明的三/二氟乙醇化试剂a(b)的应用不需要额外添加碱,以锰/过氧化 物为催化体系,可以分别与烯丙基砜,丙烯酰胺,肉桂酸反应得到三/二氟甲基高 烯丙基醇类化合物,三/二氟甲基烷基醇类化合物和三/二氟甲基烯丙基醇类化合 物。
[0067]
本发明的有益效果在于:1、本发明提供的三氟乙醇化试剂(a)和二氟乙醇 化试剂(b)可作为很多有机化合物的合成中间体,其中一些化合物具有药物活性, 简化了这类化合物的制备步骤,并且合成方法条件温和,底物适用性广;2、本发 明三氟乙醇化试剂(a)和二氟乙醇化试剂(b)的制备从简单的三氟乙醇开始, 获得对应的三氟乙酰基硅烷和二氟乙酰基硅烷,利用硼氢化钠还原得到,反应条件 温和,原料廉价易得,三氟乙醇α
‑
c
‑
si键的构建是本发明的试剂的关键,巧妙的 利用自由基brook重排后得到了三氟乙醇或者二氟乙醇切块,可以被引入其他特定 分子中,从而实现三氟乙醇化和二氟乙醇化反应。
具体实施方式
[0068]
为使本领域技术人员更好地理解本发明的技术方案,下面结合实施例对本发 明作进一步详细描述。
[0069]
实施例1
[0070]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸乙酯,合成路线及制备方法如下:
[0071][0072]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%),7a(152.4mg,0.6mmol,2.0当量),dcm(3ml,0.1 m),1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管 密封,加热至70℃搅拌18h,之后在5℃冰水浴中,加入tbaf,搅拌0.5小时后, 用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤, 经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目), 并用pe/ea(20/1~10/1,v/v)洗脱(pe:石油醚,ea:乙酸乙酯),得到53mg(产 率71%)目标化合物,为黄色油状物。对产物进行测试,测试结果如下:
[0073]
r
f
=0.23(pe/ea=8/1,v/v)。
[0074]
nmr波谱:
[0075]1h nmr(400mhz,cdcl3)δ6.35(s,1h),5.80(s,1h),4.25(q,j=7.2hz,2h), 4.16
‑
4.08(m,1h),3.63(s,1h),2.78
‑
2.57(m,2h),1.32(t,j=7.2hz,3h);
[0076]
13
c nmr(100mhz,cdcl3)δ168.1,135.2,129.8,124.9(q,j=280.5hz),70.0 (q,j=30.7hz),61.8,33.5,14.2;
[0077]
19
f nmr(375mhz,cdcl3)δ
‑
79.6(d,j=6.0hz,3f)。
[0078]
ir(atr):3444,2986,2937,1700,1633,1413,1316,1275,1163,1126,1021, 712cm
‑1。
[0079]
高分辨质谱hrms(esi,m/z):理论计算值:c8h
11
f3nao
3+
(m+na)
+
:235.0552; 发现值:235.0556。
[0080]
本实施例所用三氟乙醇化试剂1a的制备方法如下:
[0081]
1)向干燥的有磁力搅拌子的单口瓶中加入二甲基苯基氯硅烷(30mmol),三 氟乙醇(30mmol),hmpa(6ml)以及四氢呋喃溶剂后,将反应瓶置于
‑
78℃的低温 槽中,用注射泵逐滴滴加lda(二异丙基胺基锂,105mmol),滴加完毕后保持搅 拌4h,升至室温,再搅拌直至三氟乙醇消耗完毕,在0℃条件下加入三乙基氯硅烷, 搅拌4h后,经柱层析分离得到化合物4(1,1
‑
二氟
‑2‑
二甲基苯硅基
‑2‑
三乙硅氧 基乙烯);
[0082]
2)向干燥的带有磁力搅拌子的单口瓶中加入氟化剂select
‑
fluor(2.0当量) 和混合溶剂乙腈和二氯甲烷(体积比4:1),将单口瓶置于0℃冰水浴中,化合物 4加入反应中,完毕后,反应液置于室温下搅拌12小时后,用水淬灭反应,经柱 层析分离获得化合物5(三氟乙酰基苯基二甲基硅);
[0083]
3)向单口瓶中加入化合物5和甲醇,硼氢化钠固体分三批加入反应体系中, 待反应完毕后,加水淬灭反应,经柱层析分离得到三氟乙醇化试剂1a。
[0084]
三氟乙醇化试剂1a的nmr波谱:
[0085]1h nmr(400mhz,cdcl3)δ7.61(d,j=6.1hz,2h),7.47
‑
7.39(m,3h),3.84 (q,j=11.1hz,1h),0.49(d,j=3.1hz,6h);
13
c nmr(100mhz,cdcl3)δ134.4, 134.2,130.3,128.2,127.1(q,j=278.4hz),65.3(q,j=33.1hz),
‑
4.7,
‑
5.4;
19
f nmr(375mhz,cdcl3)δ
‑
70.6(d,
j=8.9hz,3f).ir(atr):3441,2963,2919, 1428,1253,1148,1085,1044,738,701cm
‑1.
[0086]
hrms(esi,m/z):理论计算值:c
10
h
13
f3nao
+
(m+na)
+
:257.0580;发现值: 257.0570。
[0087]
实施例2
[0088]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基
‑1‑
苯基戊烷
‑1‑
酮,合成路线及制备方法如下:
[0089][0090]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%),7b(257.4mg,0.9mmol,3.0当量),dcm(3ml, 0.1m),1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将 封管密封,加热至70℃搅拌14h,之后,在5℃冰水浴中,加入tbaf,搅拌0.5 小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用 盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化(200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到53mg(产率71%)目标 化合物,为黄色油状物。对产物进行测试,测试结果如下:
[0091]
r
f
=0.40(pe/ea=5/1,v/v)。
[0092]
nmr波谱:
[0093]1h nmr(400mhz,cdcl3)δ7.77(d,j=7.3hz,2h),7.60(t,j=7.3hz,1h), 7.47(t,j=7.6hz,2h),6.16(s,1h),5.90(s,1h),4.32(d,j=5.2hz,1h),4.15
‑
4.14 (m,1h),2.90
‑
2.71(m,2h);
[0094]
13
c nmr(100mhz,cdcl3)δ199.5,142.2,136.7,133.2,131.9,130.1,128.5, 125.0(q,j=280.6hz),70.5(q,j=31.2hz),33.8;
19
f nmr(375mhz,cdcl3)δ
ꢀ‑
79.3(d,j=6.0hz,3f)。
[0095]
ir(atr):3418,3064,2933,1648,1446,1338,1275,1223,1163,1036,753 cm
‑1。
[0096]
hrms(esi,m/z):理论计算值:c
12
h
12
f3o
2+
(m+h)
+
:245.0784;发现值: 245.0778。
[0097]
实施例3
[0098]1‑
([[1,1'
‑
联苯]
‑4‑
基)
‑
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊基
‑1‑
酮
[0099][0100]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%),7c(326.2mg,0.9mmol,3.0当量),dcm(3ml,0.1 m),1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,在5℃冰水浴中加入tbaf,搅拌0.5小时后,用 水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤,经 na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化(200
×
300目),并 用pe/ea(20/1~10/1,v/v)洗脱,得到74.9mg(产率78%)目标化合物,为白色固体。 对产物进行测试,测试结果如下:
[0101]
r
f
=0.50(pe/ea=5/1,v/v),沸点(mp):69
‑
71℃。
[0102]
nmr波谱:
[0103]1h nmr(400mhz,cdcl3)δ7.88(d,j=8.3hz,2h),7.69(d,j=8.3hz,2h), 7.63(d,j=7.3hz,2h),7.49(t,j=7.3hz,2h),7.42(t,j=7.2hz,1h),6.18(s,1h), 5.95(s,1h),4.38(d,j=4.9hz,1h),4.18
‑
4.17(m,1h),2.92
‑
2.74(m,2h);
[0104]
13
c nmr(100mhz,cdcl3)δ199.1,146.1,142.3,139.8,135.3,131.5,130.8, 129.1,128.5,127.4,127.2,125.0(q,j=280.8hz),70.6(q,j=30.7hz),33.9;
[0105]
19
f nmr(375mhz,cdcl3)δ
‑
79.3(d,j=6.0hz,3f)。
[0106]
ir(atr):3392,3060,2926,1640,1599,1409,1344,1275,1163,1129,1029,757 cm
‑1。
[0107]
hrms(esi,m/z):理论计算值c
18
h
16
f3o
2+
(m+h)
+
:321.1097;发现值: 321.1096。
[0108]
实施例4
[0109]1‑
(4
‑
氯苯基)
‑
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊基
‑1‑
酮
[0110]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%),7e(288.0mg,0.9mmol,3.0当量),dcm(3ml,0.1 m),1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,在5℃冰水浴中加入tbaf,搅拌0.5小时后,用 水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤,经 na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目),并 用pe/ea(20/1~10/1,v/v)洗脱,得到57.2mg(产率69%)目标化合物,为黄色油状 物。对产物进行测试,测试结果如下:
[0111]
r
f
=0.40(pe/ea=5/1,v/v)。
[0112]
nmr波谱:
[0113]1h nmr(400mhz,cdcl3)δ7.73(d,j=8.6hz,2h),7.45(d,j=8.3hz,2h), 6.16(s,1h),5.87(s,1h),4.15(s,1h),3.96(s,1h),2.90
‑
2.71(m,2h);
[0114]
13
c nmr(100mhz,cdcl3)δ198.1,142.1,139.7,135.0,131.7,131.4,128.9, 124.9(q,j=281.8hz),70.4(q,j=30.7hz),33.8;
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=8.9hz,3f)。
[0115]
ir(atr):3437,2930,1651,1588,1478,1402,1334,1275,1163,1129,1092,790 cm
‑1。
[0116]
hrms(esi,m/z):理论计算值c
12
h
11
clf3o
2+
(m+h)
+
:279.0394;发现值: 279.0389。
[0117]
实施例5
[0118]1‑
(3
‑
氯苯基)
‑
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊基
‑1‑
酮
[0119][0120]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7f(288.0mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a
(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,置于5℃冰水浴中加入tbaf,搅拌0.5小时后, 用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤, 经na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化(200
×
300目), 并用pe/ea(20/1~10/1,v/v)洗脱,得到65.1mg(产率78%)目标化合物,为黄色油 状物。对产物进行测试,测试结果如下:
[0121]
r
f
=0.40(pe/ea=5/1,v/v)。
[0122]
nmr波谱:
[0123]1h nmr(400mhz,cdcl3)δ7.73(s,1h),7.63(d,j=8.0hz,1h),7.56(d,j=8.0 hz,1h),7.41(t,j=7.8hz,1h),6.19(s,1h),5.90(s,1h),4.19
‑
4.13(m,1h),3.85(d,j =5.8hz,1h),2.91
‑
2.71(m,2h);
[0124]
13
c nmr(100mhz,cdcl3)δ197.9,142.0,138.5,134.8,133.0,132.3,129.9, 129.9,128.0,124.9(q,j=280.8hz),70.2(q,j=30.9hz),33.6;
[0125]
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=6.0hz,3f).ir(atr):3418,3071, 2930,1651,1420,1334,1275,1163,1129,1033,768cm
‑1。
[0126]
hrms(esi,m/z):理论计算值c
12
h
10
clf3nao
2+
(m+na)
+
:301.0214;发现值: 301.0216。
[0127]
实施例6
[0128]1‑
(4
‑
溴苯基)
‑
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊基
‑1‑
酮
[0129]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7h(329.0mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,置于5℃冰水浴中,加入tbaf,搅拌0.5小时后, 用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤, 经na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化(200
×
300目), 并用pe/ea(20/1~10/1,v/v)洗脱,得到78.9mg(产率81%)目标化合物,为黄色 油状物。对产物进行测试,测试结果如下:
[0130]
r
f
=0.50(pe/ea=5/1,v/v)。
[0131]
nmr波谱:
[0132]1h nmr(400mhz,cdcl3)δ7.66
‑
7.60(m,4h),6.17(s,1h),5.87(s,1h), 4.15
‑
4.14(m,1h),3.92
‑
3.91(m,1h),2.90
‑
2.71(m,2h);
[0133]
13
c nmr(100mhz,cdcl3)δ198.3,142.1,135.5,131.9,131.8,131.5,128.4, 124.9(q,j=280.8hz),70.4(q,j=30.9hz),33.7;
[0134]
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=6.0hz,3f)。
[0135]
ir(atr):3422,2920,2855,1648,1584,1398,1275,1167,1133,1074,790cm
‑1。
[0136]
hrms(esi,m/z):理论计算值c
12
h
11
brf3o
2+
(m+h)
+
:322.9889;发现值: 322.9888。
[0137]
实施例7
[0138]
5,5,5
‑
三氟
‑4‑
羟基
‑1‑
(4
‑
碘苯基)
‑2‑
亚甲基戊基
‑1‑
酮
[0139][0140]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7i(370.8mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到72.0mg(产率65%)目标 化合物,为无色油状物。对产物进行测试,测试结果如下:
[0141]
r
f
=0.50(pe/ea=5/1,v/v)。
[0142]
nmr波谱:
[0143]1h nmr(400mhz,cdcl3)δ7.83(d,j=8.3hz,2h),7.48(d,j=8.3hz,2h), 6.16(s,1h),5.86(s,1h),4.14(s,1h),3.95(d,j=4.6hz,1h),2.89
‑
2.70(m,2h);
[0144]
13
c nmr(100mhz,cdcl3)δ198.5,142.1,137.9,136.0,131.8,131.4,124.9(q,j =280.8hz),101.0,70.3(q,j=31.1hz),33.8;
[0145]
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=6.0hz,3f)。
[0146]
ir(atr):3429,2926,2855,1648,1480,1390,1275,1163,1126,1100,787cm
‑1。
[0147]
hrms(esi,m/z):理论计算值c
12
h
10
f3inao
2+
(m+na)
+
:392.9570;发现值: 392.9560。
[0148]
实施例8
[0149]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸
‑4‑
溴
‑2‑
烯
‑
丁酯
[0150][0151]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7j(323.1mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到45.7mg(产率48%)目标 化合物,为无色油状物。对产物进行测试,测试结果如下:
[0152]
r
f
=0.57(pe/ea=4/1v/v)。
[0153]
nmr波谱:
[0154]1h nmr(400mhz,cdcl3)δ6.39(s,1h),5.95
‑
5.92(m,2h),5.85(s,1h),4.71 (d,j=4.3hz,2h),4.18
‑
4.07(m,3h),3.28(s,1h),2.80
‑
2.58(m,2h);
[0155]
13
c nmr(100mhz,cdcl3)δ167.5,134.9,130.4,130.3,127.9,124.9(q,j= 281.2hz),69.9(q,j=31.2hz),64.7,43.9,33.5。
[0156]
19
f nmr(375mhz,cdcl3)δ
‑
79.6(d,j=8.9hz,3f)。
[0157]
ir(atr):3429,2922,2855,2359,2259,1715,1126,783cm
‑1。
[0158]
hrms(esi,m/z):理论计算值c
10
h
12
f3o
3+
(m
‑
br)
+
:237.0733;发现值: 237.0726。
[0159]
实施例9
[0160]
5,5,5
‑
三氟
‑1‑
(呋喃
‑2‑
基)
‑4‑
羟基
‑2‑
亚甲基戊基
‑1‑
酮
[0161][0162]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7n(248.7mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到56.0mg(产率80%)目标 化合物,为黄色油状物。对产物进行测试,测试结果如下:
[0163]
r
f
=0.40(pe/ea=5/1,v/v)。
[0164]
nmr波谱:
[0165]1h nmr(400mhz,cdcl3)δ7.71
‑
7.70(m,1h),7.26
‑
7.25(m,1h),6.59
‑
6.58(m, 1h),6.27(s,1h),6.07(s,1h),4.15
‑
4.07(m,1h),2.83
‑
2.65(m,2h);
[0166]
13
c nmr(100mhz,cdcl3)δ184.7,151.4,148.3,141.8,130.0,124.9(q,j=280.8 hz),121.9,112.6,70.5(q,j=31.2hz),34.0;
[0167]
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=6.0hz,3f)。
[0168]
ir(atr):3407,3142,2933,1618,1465,1394,1275,1163,1129,1029,768cm
‑1。
[0169]
hrms(esi,m/z):理论计算值c
10
h
10
f3o
3+
(m+h)
+
:235.0577;发现值: 235.0575。
[0170]
实施例10
[0171]
n,n
‑
二苯基5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酰胺
[0172][0173]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7z(339.3mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(8/1,v/v)洗脱,得到54mg(产率54%)目标化合物, 为无色油状物。对产物进行测试,测试结果如下:
[0174]
r
f
=0.27(pe/ea=4/1,v/v)。
[0175]
nmr波谱:
[0176]1h nmr(400mhz,cdcl3)δ7.37(t,j=7.8hz,4h),7.29
‑
7.25(m,2h), 7.19
‑
7.17(m,4h),5.45(s,1h),5.32(s,1h),4.15
‑
4.07(m,1h),2.64
‑
2.50(m,2h);
[0177]
13
c nmr(100mhz,cdcl3)δ172.4,143.2,138.6,129.5,127.3,127.2,125.8, 125.0(q,j=280.3hz),71.1(q,j=30.7hz),34.8;
19
f nmr(375mhz,cdcl3)δ
ꢀ‑
79.5(d,j=6.0hz,3f)。
[0178]
ir(atr):3288,2963,2930,1644,1592,1491,1364,1275,1163,1126,1029,693 。
[0179]
hrms(esi,m/z):理论计算值c
18
h
17
f3no
2+
(m+h)
+
:336.1206;发现值: 336.1197。
[0180]
实施例11
[0181]4‑
(5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酰基)苯腈
[0182]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7m(280.2mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到65.0mg(产率80%)目标 化合物,为黄色油状物。对产物进行测试,测试结果如下:
[0183]
r
f
=0.30(pe/ea=5/1,v/v)。
[0184]
nmr波谱:
[0185]1h nmr(400mhz,cdcl3)δ7.83(d,j=8.6hz,2h),7.77(d,j=8.6hz,2h), 6.24(s,1h),5.86(s,1h),4.23
‑
4.15(m,1h),3.42(s,1h),2.95
‑
2.74(m,2h);
[0186]
13
c nmr(100mhz,cdcl3)δ197.4,142.0,140.7,132.9,132.4,130.2,124.9(q,j =280.3hz),118.0,116.1,69.9(q,j=30.9hz),33.3;
[0187]
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=6.0hz,3f)。
[0188]
ir(atr):3448,2922,2851,2233,1655,1402,1275,1163,1129,1029,798cm
‑1。
[0189]
hrms(esi,m/z):理论计算值c
13
h
10
f3nnao
2+
(m+na)
+
:292.0556;发现值: 292.0567。
[0190]
实施例12
[0191]4‑
烯
‑4‑
苯基
‑
1,1,1
‑
三氟戊
‑2‑
醇
[0192][0193]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (29.4mg,0.12mmol,20mol%)、7ac(465.0mg,1.8mmol,3.0当量)、dcm(6ml,0.1 m)、1a(140.4mg,0.6mmol))和tbpb(291.3mg,1.5mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭
反应混合物,并用dcm(3
×
10ml)萃取。合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(30/1~20/1,v/v)洗脱,得到56mg(产率43%)目标 化合物,为黄色油状物。对产物进行测试,测试结果如下:
[0194]
r
f
=0.47(pe/ea=8/1,v/v)。
[0195]
nmr波谱:
[0196]1h nmr(400mhz,cdcl3)δ7.42
‑
7.31(m,5h),5.49(s,1h),5.28(s,1h),4.00 (bs,1h),3.11
‑
2.67(m,2h),2.21(s,1h);
[0197]
13
c nmr(100mhz,cdcl3)δ142.7,139.4,128.8,128.3,126.3,125.2(q,j=279.8 hz),121.1,117.0,68.7(q,j=30.9hz),36.3;
[0198]
19
f nmr(375mhz,cdcl3)δ
‑
79.5(d,j=6.0hz,3f)。
[0199]
ir(atr):3422,3086,3030,2960,2930,1633,1446,1390,1029,701cm
‑1。
[0200]
hrms(esi,m/z):理论计算值c
11
h
12
f3o
+
(m+h)
+
:217.0835;发现值:217.0828。
[0201]
实施例13
[0202]
表雄酮衍生物
[0203][0204]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7aj(298.8mg,0.6mmol,2.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(5/1,v/v)洗脱,得到81mg(产率59%)目标化合物, 为白色固体。对产物进行测试,测试结果如下:
[0205]
r
f
=0.24(pe/ea=2/1,v/v).mp:95
‑
97℃。
[0206]
nmr波谱:
[0207]1h nmr(400mhz,cdcl3)δ6.31(s,1h),5.77(s,1h),4.81
‑
4.73(m,1h), 4.11
‑
4.08(m,1h),3.82(s,1h),2.76
‑
2.55(m,2h),2.46
‑
2.39(m,1h),2.11
‑
2.01(m, 1h),1.95
‑
1.75(m,5h),1.67
‑
1.19(m,12h),1.09
‑
0.96(m,2h),0.86(s,3h),0.85(s, 3h),0.75
‑
0.69(m,1h);
[0208]
13
c nmr(100mhz,cdcl3)δ221.7,167.5,135.5,129.5,124.9(q,j=280.8hz), 75.1,70.0(q,j=30.9hz),54.3,51.4,47.9,44.7,36.7,36.0,35.7,35.1,33.9,33.5,31.6, 30.9,28.3,27.4,21.9,20.6,13.9,12.3;
[0209]
19
f nmr(375mhz,cdcl3)δ
‑
79.5
‑‑
79.5(m,3f,3f’)。
[0210]
ir(atr):3370,2933,2855,1718,1633,1405,1312,1291,1178,1122,1014,716 cm
‑1。
[0211]
hrms(esi,m/z):理论计算值c
25
h
35
f3nao
4+
(m+na)
+
:479.2380;发现值: 479.2370。
[0212]
实施例14
1h),5.11(s,2h),4.15
‑
4.07(m,1h),3.10(s,1h),2.78
‑
2.56(m,2h);
[0230]
13
c nmr(100mhz,cdcl3)δ167.7,148.0,147.8,135.0,130.2,129.2,124.9(q,j =280.0hz),122.5,109.1,108.4,101.4,69.9(q,j=31.1hz),67.4,33.5;
[0231]
19
f nmr(375mhz,cdcl3)δ
‑
79.6(d,j=6.0hz,3f)。
[0232]
ir(atr):3422,2900,1707,1633,1491,1446,1327,1252,1167,1122,1036,712 cm
‑1。
[0233]
hrms(esi,m/z):理论计算值c
14
h
13
f3nao
5+
(m+na)
+
:341.0607;发现值: 341.0594。
[0234]
实施例16
[0235]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸
‑4‑
溴苄酯
[0236][0237]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7v(355.5mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到60mg(56%)目标化合物, 为黄色油状物。对产物进行测试,测试结果如下:
[0238]
r
f
=0.23(pe/ea=4/1,v/v)。
[0239]
nmr波谱:
[0240]1h nmr(400mhz,cdcl3)δ7.51(d,j=8.3hz,2h),7.25(d,j=8.4hz,2h), 6.39(s,1h),5.85(s,1h),5.17(s,2h),4.17
‑
4.09(m,1h),2.81
‑
2.59(m,3h);
[0241]
13
c nmr(100mhz,cdcl3)δ167.6,134.8,134.5,132.0,130.4,130.1,124.9(q,j =279.8hz),122.7,69.9(q,j=31.1hz),66.6,33.4;
[0242]
19
f nmr(375mhz,cdcl3)δ
‑
79.6(d,j=6.0hz,3f)。
[0243]
ir(atr):3422,3528,2498,1715,1633,1439,1331,1275,1170,1126,1014,712 cm
‑1。
[0244]
hrms(esi,m/z):理论计算值c
13
h
12
brf3nao
3+
(m+na)
+
:374.9814;发现值: 374.9807。
[0245]
实施例17
[0246]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸
‑
萘
‑2‑
甲酯
[0247][0248]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7w(329.8mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a
(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,在5℃冰水浴中,加入tbaf,搅拌0.5小时后, 用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相并用盐水洗涤, 经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目), 并用pe/ea(20/1~10/1,v/v)洗脱,得到70mg(产率72%)目标化合物,为无色油 状物。对产物进行测试,测试结果如下:
[0249]
r
f
=0.3(pe/ea=10/1,v/v)。
[0250]
nmr波谱:
[0251]1h nmr(400mhz,cdcl3)δ8.03(d,j=7.9hz,1h),7.89(t,j=8.9hz,2h), 7.60
‑
7.52(m,3h),7.47(t,j=7.5hz,1h),6.35(s,1h),5.80(s,1h),5.70(s,2h), 4.17
‑
4.09(m,1h),3.31(s,1h),2.81
‑
2.59(m,2h);
[0252]
13
c nmr(100mhz,cdcl3)δ167.8,134.9,133.9,131.8,131.0,130.5,129.7, 128.95,127.87,126.86,126.19,125.40,124.9(q,j=280.8hz),123.5,69.9(q,j=30.7 hz),65.8,33.5;
[0253]
19
f nmr(375mhz,cdcl3)δ
‑
79.6(d,j=6.0hz,3f)。
[0254]
ir(atr):3444,3049,2937,1707,1633,1413,1320,1271,1167,1126,1029,775 cm
‑1。
[0255]
hrms(esi,m/z):理论计算值c
17
h
15
f3nao
3+
(m+na)
+
:347.0866;发现值: 347.0875。
[0256]
实施例18
[0257]
n,n
‑
二苯基5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酰胺
[0258][0259]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7z(339.3mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(8/1,v/v)洗脱,得到54mg(产率54%)目标化合物, 为无色油状物。对产物进行测试,测试结果如下:
[0260]
r
f
=0.27(pe/ea=4/1,v/v)。
[0261]
nmr波谱:
[0262]1h nmr(400mhz,cdcl3)δ7.37(t,j=7.8hz,4h),7.29
‑
7.25(m,2h), 7.19
‑
7.17(m,4h),5.45(s,1h),5.32(s,1h),4.15
‑
4.07(m,1h),2.64
‑
2.50(m,2h);
[0263]
13
c nmr(100mhz,cdcl3)δ172.4,143.2,138.6,129.5,127.3,127.2,125.8, 125.0(q,j=280.3hz),71.1(q,j=30.7hz),34.8;
[0264]
19
f nmr(375mhz,cdcl3)δ
‑
79.5(d,j=6.0hz,3f)。
[0265]
ir(atr):3288,2963,2930,1644,1592,1491,1364,1275,1163,1126,1029,693 cm
‑1。
[0266]
hrms(esi,m/z):理论计算值c
18
h
17
f3no
2+
(m+h)
+
:336.1206;发现值: 336.1197。
[0267]
实施例19
[0268]
n,n
‑
二甲基5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酰胺
[0269][0270]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7aa(227.9mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(10/1,v/v)洗脱,得到40mg(63%)目标化合物,为 黄色油状物。对产物进行测试,测试结果如下:
[0271]
r
f
=0.30(pe/ea=2/1,v/v)。
[0272]
nmr波谱:
[0273]1h nmr(400mhz,cdcl3)δ5.58(s,1h),5.37(s,1h),4.10
‑
4.02(m,1h),3.14(s, 3h),3.03(s,3h),2.64
‑
2.41(m,2h);
[0274]
13
c nmr(100mhz,cdcl3)δ172.4,137.6,125.1(q,j=280.3hz),121.8,71.1(q, j=30.7hz),39.8,35.5,34.8;
[0275]
19
f nmr(375mhz,cdcl3)δ
‑
79.4(d,j=6.0hz,3f)。
[0276]
ir(atr):3329,2930,1610,1454,1264,1167,1118,1029,734cm
‑1。
[0277]
hrms(esi,m/z):理论计算值c8h
12
f3nnao
2+
(m+na)
+
:234.0712;发现值: 234.0707。
[0278]
实施例20
[0279]4‑
烯
‑4‑
(4
‑
甲基苯磺酰基)
‑
1,1,1
‑
三氟
‑
戊
‑2‑
醇
[0280][0281]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (16.1mg,0.06mmol,20mol%)、7ab(302.7mg,0.9mmol,3.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌14h,之后,将schlenk管置于5℃冰水浴中,加入tbaf,搅 拌0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相 并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干,粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到63.5mg(产率72%)目标 化合物,为黄色油状物。对产物进行测试,测试结果如下:
[0282]
r
f
=0.30(pe/ea=5/1,v/v)。
[0283]
nmr波谱:
[0284]1h nmr(400mhz,cdcl3)δ7.76(d,j=8.4hz,2h),7.37(d,j=8.0hz,2h), 6.47(s,
1h),5.96(s,1h),4.30
‑
4.22(m,1h),2.63
‑
2.46(m,5h);
[0285]
13
c nmr(100mhz,cdcl3)δ145.5,145.0,134.7,130.3,128.6,128.3,124.6(q,j =277.9hz),69.1(q,j=31.4hz),31.2,21.8;
[0286]
19
f nmr(375mhz,cdcl3)δ
‑
79.7(d,j=6.0hz,3f)。
[0287]
ir(atr):3474,2930,2855,1595,1431,1279,1137,1081,734cm
‑1。
[0288]
hrms(esi,m/z):理论计算值c
12
h
13
f3nao3s
+
(m+na)
+
:317.0430;发现值: 317.0432。
[0289]
实施例21
[0290]3‑
甲基
‑1‑
苯基
‑3‑
(3,3,3
‑
三氟
‑2‑
羟丙基)吲哚
‑2‑
酮
[0291][0292]
在氮气保护下,向干燥的含有合适大小聚四氟乙烯磁子的25ml schlenk反应管 中分别加入mn(oac)2·
4h2o(29.4mg,0.12mmol,20mol%)、10a(170.7mg,0.72 mmol,1.2当量),然后继续加入dcm(6ml,0.1m)、1a(140.4mg,0.6mmol)和 tbpb(291.5mg,1.5mmol,2.5当量),将反应管密封,然后将反应管放在加热模块上 加热到70℃反应14h,取下反应管,待反应管冷却到室温,将反应管放入到冰水浴 中,打开反应管,加入tbaf(188.3mg,0.72mmol,1.2当量),然后将反应管密封, 将反应在冰水浴中搅拌30分钟,再用8ml的水将反应淬灭,用饱和氯化钠(30ml) 和dcm(3
×
20ml)对反应进行萃取,合并有机相,然后用饱和氯化钠(2
×
50ml)洗 涤有机相,用无水na2so4干燥有机相,过滤有机相,用减压旋转蒸发仪浓缩得到粗 产物,所得粗产物用石油醚和乙酸乙酯为洗脱剂,通过硅胶柱层析分离得到3
‑
甲基
‑1‑ꢀ
苯基
‑3‑
(3,3,3
‑
三氟
‑2‑
羟丙基)吲哚
‑2‑
酮(两个非对应异构体总产率91%)。对产 物进行测试,测试结果如下:
[0293]
大极性非对应异构体:r
f
=0.32(pe/ea=5/1v/v)。mp:116
‑
118℃。
[0294]
nmr波谱:
[0295]1h nmr(400mhz,cdcl3)δ7.54
–
7.49(m,2h),7.43
–
7.39(m,3h),7.26
–
7.22(m, 2h),7.16
–
7.12(m,1h),6.85
–
6.82(m,1h),3.66
–
3.57(m,1h),2.53
‑
2.22(m,2h),1.91 (s,1h),1.54(s,3h)。
[0296]
13
c nmr(150mhz,cdcl3)δ180.4,143.8,134.6,131.6,129.8,128.6,128.3, 126.8,124.8(q,j=282.2hz),123.4,122.9,110.0,68.7(q,j=31.4hz),46.1,37.8, 25.8。
[0297]
19
f nmr(375mhz,cdcl3)δ
‑
80.0(d,j=7.2hz,3f)。
[0298]
ir(atr):3377,3056,2967,2926,1703,1610,1506,1379,1282,1163,1126,1028, 854,760cm
‑1。
[0299]
hrms(esi,m/z):理论计算值c
18
h
16
f3no2na
+
(m+na)
+
:358.1025;发现值: 358.1012。
[0300]
实施例22
[0301]
(e)
‑
1,1,1
‑
三氟
‑4‑
(2
‑
氟苯基)
‑3‑
烯
‑
丁
‑2‑
醇
[0302][0303]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)3·
2h2o (21.4mg,0.08mmol,20mol%)、12a(132.8mg,0.8mmol,2.0当量)、正己烷(1ml,0.4 m)、1a(93.6mg,0.4mmol)和tbpb(194.3mg,1.0mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,将schlenk管置于
‑
10℃低温槽中,加入tbaf, 搅拌1.0小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机 相并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目),并用pe/ea(20/1,v/v)洗脱,得到64mg(产率73%)目标化合物, 为白色固体状物。对产物进行测试,测试结果如下:
[0304]
r
f
=0.56(pe/ea=4/1,v/v)。
[0305]
nmr波谱:
[0306]1h nmr(400mhz,cdcl3)δ7.47(t,j=7.2hz,1h),7.31
‑
7.26(m,1h), 7.15
‑
7.00(m,3n),6.31(dd,j=16.2,6.4hz,1h),4.69
‑
4.63(m,1h),2.44(s,1h);
[0307]
13
c nmr(100mhz,cdcl3)δ160.7(d,j=250.5hz),130.3(d,j=8.7hz),129.0 (d,j=2.9hz),128.1(d,j=2.9hz),124.4(q,j=273.4hz),124.4(d,j=3.9hz), 123.4,123.4(d,j=6.7hz),116.1(d,j=22.2hz),71.9(q,j=32.1hz);
[0308]
19
f nmr(375mhz,cdcl3)δ
‑
78.9(d,j=6.0hz,3f),
‑
117.0
‑‑
117.0(m,1f)。
[0309]
ir(atr):3396,2922,1659,1491,1457,1267,1174,1125,969,883,753cm
‑1。
[0310]
rms(esi,m/z):理论计算值c
10
h6f4o
‑
(m
‑
h)
‑
:219.0439;发现值:219.0441。
[0311]
实施例23
[0312]
5,5
‑
二氟
‑1‑
苯基
‑4‑
羟基
‑2‑
亚甲基戊烷
‑1‑
酮
[0313][0314]
在氮气保护下,向干燥的含有合适大小聚四氟乙烯磁子的10ml schlenk反应管 中分别加入mn(oac)3·
2h2o(16.1mg,0.06mmol,20mol%)、7b(257.4mg,0.9mmol, 3.0当量),然后继续加入dcm(3ml,0.1m)、2a(64.8mg,0.3mmol)和tbpb (145.7mg,0.75mmol,2.5当量),将反应管密封,然后将反应管放在加热模块上加热 到70℃反应14h,取下反应管,待反应管冷却到室温,将反应管放入到冰水浴中, 打开反应管,加入tbaf(1.0m in thf,0.36ml,0.36mmol,1.2当量),然后将反应 管密封,将反应管在冰水浴中搅拌30分钟。然后用2ml的水将反应淬灭,dcm(3
×
10 ml)对反应液进行萃取,合并有机相,然后用饱和氯化钠(2
×
25ml)洗涤有机相, 用无水na2so4干燥有机相,过滤有机相,用减压旋转蒸发仪浓缩得到粗产物,所得 粗产物用石油醚和乙酸乙酯为洗脱剂,通过硅胶柱层析分离得到无色液体的5,5
‑
二氟
ꢀ‑1‑
苯基
‑4‑
羟基
‑2‑
亚甲基戊烷
‑1‑
酮(产率80%)。对产物进行测试,测试结果如下:
[0315]
r
f
=0.25(pe/ea=10/1,v/v)。
[0316]
nmr波谱:
[0317]1h nmr(400mhz,cdcl3)δ7.77(d,j=7.3hz,2h),7.58(t,j=7.3hz,1h),7.45 (t,j=7.6hz,2h),6.11(s,1h),5.86
‑
5.58(m,2h),3.98
‑
3.91(m,1h),3.83(s,1h), 2.84
‑
2.63(m,2h);
[0318]
13
c nmr(100mhz,cdcl3)δ199.5,143.1,137.0,133.0,130.9,130.0,128.5, 116.1(t,j=244.2hz),70.9(t,j=24.1hz),33.7;
[0319]
19
f nmr(375mhz,cdcl3)δ
–
128.5
––
131.2(m,2f)。
[0320]
ir(atr):3452,3064,2941,2292,2251,1655,1446,1409,1375,1330,1219,1174, 1140,1059,947,757cm
‑1。
[0321]
hrms(esi,m/z):理论计算值c
12
h
13
f2o
2+
(m+h)
+
:227.0878;发现值:227.0871。
[0322]
本实施例所用二氟乙醇化试剂2a的制备方法为:
[0323]
1)向带有磁力搅拌子的单口瓶中加入30mmol化合物4(同实施例1中化合物4) 和溶剂thf,并置于0℃冰浴中,向反应液中滴加浓盐酸(10当量),滴加完毕后,将 反应装置移至室温条件下继续搅拌,反应完全后,加水淬灭反应,经柱层析分离得 到化合物6。
[0324]
2)向带有磁力搅拌子的单口瓶中加入10mmol化合物6和溶剂甲醇(0.2m), 并置于0℃冰浴中,硼氢化钠固体(1.1当量)分三批加入反应体系中,反应完全后, 加水淬灭反应,经柱层析分离得到二氟乙醇化试剂2a。
[0325]
二氟乙醇化试剂2a的nmr波谱:
[0326]1h nmr(400mhz,cdcl3)δ7.57(d,j=6.4hz,2h),7.45
‑
7.39(m,3h),5.39(t,j =54.9hz,1h),0.62(s,6h);
[0327]
13
c nmr(100mhz,cdcl3)δ233.0(t,j=32.0hz),134.3,132.4,130.5,128.4, 112.2(t,j=249.9hz),
‑
4.7;
[0328]
19
f nmr(375mhz,cdcl3)δ
‑
125.3(d,j=53.6hz,3f)。
[0329]
ir(atr):3071,2960,1670,1428,1252,1118,1044,828,787,697cm
‑1。
[0330]
hrms(esi,m/z):理论计算值:c
10
h
13
f2osi
+
(m+h)
+
:215.0698;发现值: 215.0693。
[0331]
实施例24
[0332]
薯蓣皂素二氟衍生物
[0333][0334]
在氮气保护下,向干燥的含有合适大小聚四氟乙烯磁子的10ml schlenk反应管 中分别加入mn(oac)2·
4h2o(14.7mg,0.06mmol,20mol%)、7am(373.5mg,0.6mmol, 2.0当量),然后继续加入dcm(3ml,0.1m)、2a(64.8mg,0.3mmol)和tbpb(145.7 mg,0.75mmol,2.5当量)。将反应管密封,然后将反应管放在加热模块上加热到70℃ 反应18h,取下反应管,待反应管冷却到室温,将反应管放入到冰水浴中,打开反 应管,加入tbaf(1.0m in thf,0.36ml,0.36mmol,1.2当量),然后将反应管密封, 将反应管在冰水浴中搅拌30分钟。然后用2ml的水将反应淬灭,dcm(3
×
10ml) 对反应液进行萃取,合并有机相,然后用饱和氯化钠(2
×
25ml)洗涤有机相,用无 水na2so4干燥有机相,过滤有机相,用减压旋转蒸发仪浓缩得
到粗产物,所得粗产 物用石油醚和乙酸乙酯为洗脱剂,通过硅胶柱层析分离得到白色固体的薯蓣皂素二 氟衍生物(产率67%)。对产物进行测试,测试结果如下:
[0335]
r
f
=0.42(pe/ea=5/1,v/v)。mp:126.1
‑
127.4℃。
[0336]
nmr波谱:
[0337]1h nmr(400mhz,cdcl3)δ6.29(d,j=1.3hz,1h),5.84
–
5.52(m,2h),5.39(d,j =5.1hz,1h),4.74
–
4.61(m,1h),4.40(dd,j=14.3,8.1hz,1h),3.98
–
3.84(m,1h), 3.50
–
3.43(m,1h),3.36(t,j=10.9hz,1h),2.69(dd,j=14.4,3.3hz,1h),2.51(dd,j =14.4,9.0hz,1h),2.37(d,j=7.9hz,2h),2.06
–
1.40(m,18h),1.02(s,3h),0.96(d,j =7.0hz,3h),0.78(d,j=4.8hz,6h);
[0338]
13
c nmr(100mhz,cdcl3)δ167.4,139.5,136.3,122.8,116.0(t,j=244.3hz), 109.4,80.9,75.3,70.6(t,j=23.8hz),67.0,62.2,56.6,41.7,40.4,39.8,38.1,37.0,36.9, 33.4(t,j=4.0hz),32.2,32.0,31.5,28.9,27.8,20.9,19.5,17.2,16.4,14.6;
[0339]
19
f nmr(375mhz,cdcl3)δ
–
128.7
––
131.6(m,2f)。
[0340]
ir(atr):3418,2945,1710,1454,1375,1327,1245,1051,980,83cm
‑1。
[0341]
hrms(esi,m/z):理论计算值c
33
h
48
f3o
5+
(m+h)
+
:563.3543;发现值:563.3533。
[0342]
实施例25
[0343]3‑
(3,3
‑
二氟
‑2‑
羟丙基)
‑3‑
甲基
‑2‑
氧
‑1‑
苯基二氢吲哚
‑6‑
羧酸甲酯
[0344][0345]
在氮气保护下,向干燥的含有合适大小聚四氟乙烯磁子的10ml schlenk反应管 中分别加入mn(oac)3·
2h2o(16.1mg,0.06mmol,20mol%)、10i(83.4mg,0.36mmol, 1.2当量),然后继续加入dcm(3ml,0.1m)、2a(64.8mg,0.3mmol)和tbpb(145.7 mg,0.75mmol,2.5当量)。将反应管密封,然后将反应管放在加热模块上加热到70℃ 反应14h,取下反应管,待反应管冷却到室温,将反应管放入到冰水浴中,打开反 应管,加入tbaf(1.0m in thf,0.36ml,0.36mmol,1.2当量),然后将反应管密封, 将反应管在冰水浴中搅拌30分钟。然后用2ml的水将反应淬灭,dcm(3
×
10ml) 对反应液进行萃取,合并有机相,然后用饱和氯化钠和碳酸钠洗涤有机相,用无水 na2so4干燥有机相,过滤有机相,用减压旋转蒸发仪浓缩得到粗产物,所得粗产物 用石油醚和乙酸乙酯为洗脱剂,通过硅胶柱层析分离得到3
‑
(3,3
‑
二氟
‑2‑
羟丙基)
‑3‑ꢀ
甲基
‑2‑
氧
‑1‑
苯基二氢吲哚
‑6‑
羧酸甲酯(产率76%,14h
‑
a:14h
‑
b=51:49)。对产 物进行测试,测试结果如下:
[0346]
r
f
(14h
‑
a)=0.28(pe/ea=2/1,v/v).(36.8mg,39%yield,白色固体,mp: 107.2
–
108.9℃。
[0347]
14h
‑
a的nmr波谱:
[0348]1h nmr(400mhz,cdcl3)δ8.05(dd,j=8.1,1.7hz,1h),7.88(s,1h),6.92(d,j =8.2hz,1h),5.81
–
5.52(m,1h),4.44(s,1h),4.19
–
4.01(m,1h),3.91(s,3h),3.28(s, 3h),2.18
–
1.80(m,2h),1.49(s,3h);
[0349]
13
c nmr(100mhz,cdcl3)δ182.4,166.8,146.4,134.8,131.2,125.5,123.9, 116.0
(t,j=240.6hz),108.4,68.6(t,j=24.1hz),52.3,46.5,36.0,26.9,22.6;
[0350]
19
f nmr(375mhz,cdcl3)δ
–
124.8
––
133.7(m,2f)。
[0351]
ir(atr):3414,2926,1703,1498,1457,1286,1103,1051,977,772cm
‑1。
[0352]
hrms(esi,m/z):理论计算值c
15
h
18
f2no
4+
(m+h)
+
:314.1198;发现值: 314.1198。
[0353]
r
f
(14h
‑
b)=0.17(pe/ea=2/1,v/v).(34.5mg,37%yield,白色固体,mp: 128.4
‑
129.8℃)。
[0354]
14h
‑
b的nmr波谱:
[0355]1h nmr(400mhz,cdcl3)δ8.05(dd,j=8.1,1.7hz,1h),6.92(d,j=8.2hz, 1h),5.81
–
5.52(m,1h),4.43(s,1h),4.21
–
4.07(m,1h),3.91(s,3h),3.27(s,3h), 2.23
–
1.76(m,1h),1.49(s,3h);
[0356]
13
c nmr(100mhz,cdcl3)δ181.6,167.0,147.8,132.5,131.2,124.7,124.0, 115.8(t,j=242.5hz),108.1,68.8(t,j=23.8hz),52.2,45.9,37.2,26.7,25.2;
[0357]
19
f nmr(375mhz,cdcl3)δ
–
126.5
––
133.0(m,2f)。
[0358]
ir(atr):3422,2922,1707,1498,1457,1372,1286,1055,977,772cm
‑1。
[0359]
hrms(esi,m/z):理论计算值c
15
h
18
f2no
4+
(m+h)
+
:314.1198;发现值: 314.1197。
[0360]
实施例26
[0361]
(e)
‑4‑
(2,6
‑
二氟苯基)
‑
1,1
‑
二氟丁
‑3‑
烯
‑2‑
醇
[0362][0363]
在氮气保护下,向干燥的含有合适大小聚四氟乙烯磁子的10ml schlenk反应管 中分别加入mn(oac)3·
2h2o(16.1mg,0.06mmol,20mol%)、12d(110.4mg,0.6mmol, 2.0当量),然后继续加入dcm(3ml,0.1m)、2a(64.8mg,0.3mmol)和tbpb(145.7 mg,0.75mmol,2.5当量)。将反应管密封,然后将反应管放在加热模块上加热到70℃ 反应14h,取下反应管,待反应管冷却到室温,将反应管放入到冰水浴中,打开反 应管,加入tbaf(1.0m in thf,0.36ml,0.36mmol,1.2当量),然后将反应管密封, 将反应在冰水浴中搅拌30分钟。然后用2ml的水将反应淬灭,dcm(3
×
10ml)对反 应液进行萃取,合并有机相,然后用饱和氯化钠和碳酸钠洗涤有机相,用无水na2so4干燥有机相,过滤有机相,用减压旋转蒸发仪浓缩得到粗产物,所得粗产物用石油 醚和乙酸乙酯为洗脱剂,通过硅胶柱层析分离得到浅黄色液体(e)
‑4‑
(2,6
‑
二氟苯基)
ꢀ‑
1,1
‑
二氟丁
‑3‑
烯
‑2‑
醇(产率60%)。对产物进行测试,测试结果如下:
[0364]
r
f
=0.40(pe/ea=5/1,v/v)。
[0365]
nmr波谱:
[0366]1h nmr(400mhz,cdcl3)δ7.24
–
7.16(m,1h),6.94
–
6.84(m,3h),6.59
–
6.53(m, 1h),5.89
–
5.59(m,1h),4.48(dq,j=10.3,5.2hz,1h),2.23(s,1h);
[0367]
13
c nmr(150mhz,cdcl3)δ161.2(dd,j=250.3,7.3hz),129.7
–
129.4(m), 129.2(t,j=10.8hz),121.2,115.5(t,j=243.8hz),113.4(t,j=15.1hz),111.7(dd,j =21.5,4.8hz),72.8(t,j=24.4hz);
[0368]
19
f nmr(375mhz,cdcl3)δ
–
112.7(s,2f),
–
126.3
––
130.0(m,2f)。
[0369]
ir(atr):3396,2926,1621,1584,1464,1267,1118,1062,999,909cm
‑1。
[0370]
hrms(esi,m/z):理论值c
10
h8f4ona
+
(m+na)
+
:243.0404;发现值:243.0412。
[0371]
实施例27
[0372]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸
‑5‑
醛基戊酯
[0373][0374]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7a(152.4mg,0.6mmol,2.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,将schlenk管置于5℃冰水浴中加入tbaf,搅拌 0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相并 用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(20/1~10/1,v/v)洗脱,得到39mg(46%)目标化合物, 为无色油状物。对产物进行测试,测试结果如下:
[0375]
r
f
=0.61(pe/ea=2/1,v/v)。
[0376]
nmr波谱:
[0377]1h nmr(400mhz,cdcl3)δ9.77(t,j=1.5hz,1h),6.33(s,1h),5.80(s,1h), 4.22
‑
4.09(m,3h),2.79
‑
2.57(m,2h),2.47(td,j=7.2,1.3hz,2h),1.76
‑
1.64(m,4h), 1.46
‑
1.38(m,2h);
[0378]
13
c nmr(100mhz,cdcl3)δ202.5,167.9,135.2,129.8,124.9(q,j=281.1hz), 69.9(q,j=30.7hz),65.3,43.8,33.5,28.4,25.6,21.7;
[0379]
19
f nmr(375mhz,cdcl3)δ
‑
79.6(d,j=6.0hz,3f)。
[0380]
ir(atr):3425,2930,2859,1711,1275,1167,1126,1029,734cm
‑1。
[0381]
hrms(esi,m/z):理论计算值c
12
h
17
f3nao
4+
(m+na)
+
:305.0971;发现值: 305.0970。
[0382]
实施例28
[0383]
胆固醇衍生物
[0384][0385]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%)、7ak(357.6mg,0.6mmol,2.0当量)、dcm(3ml,0.1 m)、1a(70.2mg,0.3mmol))和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密 封,加热至70℃搅拌18h,之后,将schlenk管置于5℃冰水浴中加入tbaf,搅拌 0.5小时后,用水(2ml)淬灭反应混合物,并用dcm(3
×
10ml)萃取。合并有机相并 用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化 (200
×
300目),并用pe/ea(5/1,v/v)洗脱,得到53mg(71%)目标化合物,为黄 色油状物。对产物进行测试,测试结果如下:
[0386]
r
f
=0.48(pe/ea=8/1,v/v).mp:83
‑
85℃。
[0387]
nmr波谱:
[0388]1h nmr(400mhz,cdcl3)δ6.33(s,1h),5.78(s,1h),5.40(d,j=4.3hz,1h), 4.73
‑
4.65(m,1h),4.12
‑
4.09(m,1h),3.75(s,1h),2.77
‑
2.57(m,2h),2.37(d,j=7.6 hz,2h),2.03
‑
1.95(m,2h),1.92
‑
1.79(m,3h),1.70
‑
1.43(m,7h),1.40
‑
1.25(m,4h), 1.20
‑
1.08(m,7h),1.03
‑
0.95(m,6h),0.91(d,j=6.4hz,3h),0.86(dd,j=6.7,1.8hz, 6h),0.68(s,3h);
[0389]
13
c nmr(100mhz,cdcl3)δ167.6,139.4,135.6,129.6,124.9(q,j=280.8hz), 123.2,75.64,70.1(q,j=30.7hz),56.8,56.2,50.1,42.4,39.8,39.6,38.1,37.0,36.7, 36.3,35.9,33.6,32.0,31.9,28.4,28.1,27.8,24.4,24.0,23.0,22.7,21.2,19.5,18.8, 12.0;
[0390]
19
f nmr(375mhz,cdcl3)δ
‑
79.5(bs,3f)。
[0391]
ir(atr):3422,2937,2870,1711,1633,1465,1331,1275,1170,1129,1029,734 cm
‑1。
[0392]
hrms(esi,m/z):理论计算值c
33
h
51
f3nao
3+
(m+na)
+
:575.3683;发现值: 575.3671。
[0393]
实施例29
[0394]
根据三氟乙醇化试剂1a或二氟乙醇化试剂2a合成具有抗癌活性的化合物l (1,1,1
‑
三氟
‑3‑
烯
‑4‑
(2
‑
甲基
‑3‑
(3
‑
(3,4
‑
二羟甲基)苯基)丙基)苯基丁
‑2‑
醇) 及其二氟类似物m(1,1
‑
二氟
‑3‑
烯
‑4‑
(2
‑
甲基
‑3‑
(3
‑
(3,4
‑
二羟甲基)苯基)丙基) 苯基丁
‑2‑
醇)的方法,合成方法反应方程式可如下表示:
[0395][0396]
所述r
f
为cf3–
或hcf2–
,r
f
为cf3–
时,所得产物为具有抗癌活性的化合物 l,r
f
为hcf2–
时,所得产物为l的二氟类似物m。
[0397]
具体地,其合成方法包含以下步骤:
[0398]
1)在氮气保护下,反应物24和三乙胺(1.4当量)溶解在溶剂dcm中,反 应置于
‑
78℃的低温浴中,缓慢滴加三氟甲磺酸酐(1.1当量),滴加完毕后,饱和 氯化铵水溶液淬灭反应,dcm萃取,柱层析获得化合物25;
[0399]
2)化合物25和叔丁基丙烯酸酯(5当量)在醋酸钯(化合物25摩尔量的20%) 和dppp(1,3
‑
双(二苯膦)丙烷,化合物25摩尔量的22%)为催化体系,三乙胺(3.0 当量)作为碱,dmf为溶剂的反应体系中,110℃的温度下搅拌12h,加水淬灭反 应,乙酸乙酯萃取,经柱层析得到的化合物在溶剂二氯甲烷中经三氟乙酸处理,得 到化合物26;
[0400]
3)利用化合物26和三氟乙醇化试剂1a或者二氟乙醇化试剂2a反应,在氮 气保护下,向干燥的含有合适大小聚四氟乙烯磁子的10ml schlenk反应管中分别 加入mn(oac)3·
2h2o(16.1mg,0.06mmol,20mol%)、26(0.6mmol,2.0当量),然 后继续加入dcm
(3ml,0.1m)、1a或2a(0.3mmol)和tbpb(145.7mg,0.75mmol, 2.5当量),将反应管密封,然后将反应管放在加热模块上加热到70℃反应14h, 取下反应管,待反应管冷却到室温,将反应管放入到冰水浴中,打开反应管,加入 tbaf(1.0m in thf,0.36ml,0.36mmol,1.2当量),然后将反应管密封,将反应在 冰水浴中搅拌30分钟。然后用2ml的水将反应淬灭,dcm萃取(3
×
10ml),经 柱层析得到目标产物27或者化合物28;
[0401]
4)化合物27或者化合物28溶解在二氯甲烷溶剂中,反应置于
‑
78℃低温槽 中,缓慢滴加dibal
‑
h(二异丁基氢化铝),滴加完毕后缓慢升至0℃,用3m hcl 淬灭反应,经柱层析得到抗癌活性化合物l或二氟类似物m。
[0402]
实施例30
[0403]
5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸乙酯,合成路线及制备方法如下:
[0404][0405]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%),7a(152.4mg,0.6mmol,2.0当量),dcm(3ml,0.1 m),a2(0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密封,加 热至70℃搅拌18h,之后在5℃冰水浴中,加入tbaf,搅拌0.5小时后,用水(2ml) 淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目),并用pe/ea (20/1~10/1,v/v)洗脱(pe:石油醚,ea:乙酸乙酯),得到35mg(产率47%)
[0406]
本实施例所用三氟乙醇化试剂a2的制备方法如下:
[0407]
1)向干燥的有磁力搅拌子的单口瓶中加入三乙基氯硅烷(30mmol),三氟 乙醇(30mmol)以及四氢呋喃溶剂后,将反应瓶置于
‑
78℃的低温槽中,用注射泵 逐滴滴加lda(二异丙基胺基锂,105mmol),滴加完毕后保持搅拌4h,升至室 温,再搅拌直至三氟乙醇消耗完毕,在0℃条件下加入三乙基氯硅烷,搅拌4h后, 经柱层析分离得到化合物4(1,1
‑
二氟
‑2‑
三乙基硅基
‑2‑
三乙硅氧基乙烯);
[0408]
2)向干燥的带有磁力搅拌子的单口瓶中加入氟化剂select
‑
fluor(2.0当量) 和混合溶剂乙腈和二氯甲烷(体积比4:1),将单口瓶置于0℃冰水浴中,化合物4加入反应中,完毕后,反应液置于室温下搅拌12小时后,用水淬灭反应,经柱 层析分离获得化合物5(三氟乙酰基三乙基硅);
[0409]
3)向单口瓶中加入化合物5和甲醇,硼氢化钠固体分三批加入反应体系中, 待反应完毕后,加水淬灭反应,经柱层析分离得到三氟乙醇化试剂a2。
[0410]
实施例31
[0411]
以a3为原料,5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸乙酯,合成路线及制备方法如 下:
[0412][0413]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%),7a(152.4mg,0.6mmol,2.0当量),dcm(3ml,0.1 m),a3(0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密封,加 热至70℃搅拌18h,之后在5℃冰水浴中,加入tbaf,搅拌0.5小时后,用水(2ml) 淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目),并用pe/ea (20/1~10/1,v/v)洗脱(pe:石油醚,ea:乙酸乙酯),得到25.4mg(产率34%)。
[0414]
本实施例所用三氟乙醇化试剂a3的制备方法如下:
[0415]
1)向干燥的有磁力搅拌子的单口瓶中加入三苯基氯硅烷(30mmol),三氟 乙醇(30mmol),hmpa(3ml)以及四氢呋喃溶剂后,将反应瓶置于
‑
78℃的低 温槽中,用注射泵逐滴滴加lda(二异丙基胺基锂,105mmol),滴加完毕后保 持搅拌4h,升至室温,再搅拌直至三氟乙醇消耗完毕,在0℃条件下加入三乙基 氯硅烷,搅拌4h后,经柱层析分离得到化合物4(1,1
‑
二氟
‑2‑
三苯基硅基
‑2‑
三乙 硅氧基乙烯);
[0416]
2)向干燥的带有磁力搅拌子的单口瓶中加入氟化剂select
‑
fluor(2.0当量) 和混合溶剂乙腈和二氯甲烷(体积比4:1),将单口瓶置于0℃冰水浴中,化合物 4加入反应中,完毕后,反应液置于室温下搅拌12小时后,用水淬灭反应,经柱 层析分离获得化合物5(三氟乙酰基三苯基硅);
[0417]
3)向单口瓶中加入化合物5和甲醇,硼氢化钠固体分三批加入反应体系中, 待反应完毕后,加水淬灭反应,经柱层析分离得到三氟乙醇化试剂a3。
[0418]
实施例32
[0419]
以a4为原料,5,5,5
‑
三氟
‑4‑
羟基
‑2‑
亚甲基戊酸乙酯,合成路线及制备方法如 下:
[0420][0421]
在氮气气氛下,向干燥的10ml schlenk管中放入磁子,加入mn(oac)2·
4h2o (14.7mg,0.06mmol,20mol%),7a(152.4mg,0.6mmol,2.0当量),dcm(3ml,0.1 m),a4(0.3mmol)和tbpb(145.7mg,0.75mmol,2.5当量),然后将封管密封,加 热至70℃搅拌18h,之后在5℃冰水浴中,加入tbaf,搅拌0.5小时后,用水(2ml) 淬灭反应混合物,并用dcm(3
×
10ml)萃取,合并有机相并用盐水洗涤,经na2so4干燥,使用旋转蒸发仪悬干。粗产物经硅胶柱色谱纯化(200
×
300目),并用pe/ea (20/1~10/1,v/v)洗脱(pe:石油醚,ea:乙酸乙酯),得到35.8mg(产率48%)
[0422]
本实施例所用三氟乙醇化试剂a4的制备方法如下:
[0423]
1)向干燥的有磁力搅拌子的单口瓶中加入三苯基氯硅烷(30mmol),三氟 乙醇
(30mmol),hmpa(3ml)以及四氢呋喃溶剂后,将反应瓶置于
‑
78℃的低 温槽中,用注射泵逐滴滴加lda(二异丙基胺基锂,105mmol),滴加完毕后保 持搅拌4h,升至室温,再搅拌直至三氟乙醇消耗完毕,在0℃条件下加入三乙基 氯硅烷,搅拌4h后,经柱层析分离得到化合物4(1,1
‑
二氟
‑2‑
甲基二苯硅基
‑2‑
三 乙硅氧基乙烯);
[0424]
2)向干燥的带有磁力搅拌子的单口瓶中加入氟化剂select
‑
fluor(2.0当量) 和混合溶剂乙腈和二氯甲烷(体积比4:1),将单口瓶置于0℃冰水浴中,化合物 4加入反应中,完毕后,反应液置于室温下搅拌12小时后,用水淬灭反应,经柱 层析分离获得化合物5(三氟乙酰基甲基二苯基硅);
[0425]
3)向单口瓶中加入化合物5和甲醇,硼氢化钠固体分三批加入反应体系中, 待反应完毕后,加水淬灭反应,经柱层析分离得到三氟乙醇化试剂a4。
[0426]
实施例33
[0427]
测试本申请实施例1、实施例23、实施例30、实施例31、实施例32制备的 三(二)氟乙醇化试剂1a、2a、a2、a3、a4的储存稳定性、光稳定性及在常见 溶剂中的溶解性,结果见下表1。
[0428]
表1
[0429]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1